Niemann-Pick disease is a rare lysosomal storage disorder inherited in an autosomal recessive manner, which disrupts the body’s ability to metabolize fats, resulting in their accumulation within cells. This disorder can affect various organs, most commonly the brain, liver, spleen, bone marrow, and lungs. The defining features include hepatosplenomegaly, failure to thrive, and neurological deficits.
The three main types of Niemann-Pick disease are NPD-A (type A), NPD-B (type B), and NPD-C (type C). NPD-A and NPD-B result from a deficiency in the enzyme acid sphingomyelinase caused by mutations in the SMPD1 gene. Mutations in the NPC1 and NPC2 genes cause NPD-C.
This report discusses an 11-month-old infant who presented to the outpatient department with failure to thrive, abdominal distension, and developmental delay. On examination, the infant was emaciated, pale, and exhibited hepatosplenomegaly and developmental delay. Bone marrow and liver biopsies revealed characteristic lipid-laden foamy macrophages. Detailed history, examination, and investigations confirmed a diagnosis of NPD-A.
NPD-A has a poor prognosis and is usually fatal by three years of age. The patient received supportive treatment, including nutritional therapy and physiotherapy, and the parents were counseled regarding the disease outcome. The patient has been regularly followed up, with two episodes of chest infections reported during an eight-month follow-up period.
Introduction: Niemann-Pick Disease Type A
Niemann-Pick disease (NPD) is a lysosomal storage disorder inherited in an autosomal recessive manner. It affects lipid metabolism, particularly sphingomyelin, a fat found in cell membranes. This results in the accumulation of sphingomyelin in various organs and tissues, leading to a range of symptoms and complications. NPD is classified into three main types: NPD-A, NPD-B, and NPD-C, each with distinct symptoms and progression rates.
NPD-A and NPD-B are caused by a deficiency in the enzyme acid sphingomyelinase due to missense mutations in the SMPD1 gene (sphingomyelin phosphodiesterase 1). NPD-C results from mutations in the NPC1 and NPC2 genes, which encode proteins responsible for lysosomal cholesterol transport.
NPD-A is a fatal infantile disorder characterized by failure to thrive, hepatosplenomegaly, and a rapidly progressing neurodegenerative course, often leading to death by the age of three [1]. The incidence of NPD-A is approximately 1 in 250,000 in the general population but is more prevalent among Ashkenazi Jews, with an estimated incidence of 1 in 40,000 [2-5].
NPD-B is a non-neuropathic form, less severe with a later onset, affecting both children and adults. It is characterized by the accumulation of sphingomyelin in non-neuronal tissues, including the lungs, leading to progressive lung disease. At diagnosis, NPD-B typically presents with reticular or finely nodular infiltrations visible on a chest X-ray and has a better prognosis [2-4].
NPD-C, the most common neuropathic form, is caused by mutations in the NPC1 and NPC2 genes. It typically begins around two years but can also appear in adults. Ataxia, quadriparesis, seizures, and moderate hepatosplenomegaly characterize NPD-C [2-4,6].
A patient with NPD-A typically appears normal at birth. By six months of age, symptoms such as hepatosplenomegaly and developmental delay become apparent. As the disease progresses, neuroregression occurs, leading to the loss of developmental milestones like social smiling, neck control, and sitting. This is followed by a decline in intellectual abilities and the onset of spasticity [2-5].
Case Presentation
Patient Profile
- An 11-month-old male infant presented to the OPD of Lady Reading Hospital Peshawar with the following chief complaints:
- Failure to thrive
- Gradual abdominal distension since the 5th month of ag
- Developmental delay
Background Information
- The infant is the 2nd issue of a first-degree consanguineous marriage.
- Born via spontaneous vaginal delivery at the 37th week of gestation with a normal birth weight of 3.5 kg.
- Past medical history includes multiple hospital admissions for recurrent chest infections.
- Developmental history indicates delayed milestones:
- A social smile started at the 5th month of age
- Neck holding at the 6th month
- Sitting with support at the 7th month
- Regression of milestones began after the 7th month.
Medical History
- Positive transfusion history of packed red blood cells at eight months due to severe iron deficiency anemia.
- Vaccination history is up-to-date.
General Physical Examination
- Emaciated and pale appearance.
- Coarse facies with nasal bridge depression and a prominent forehead (Figure 1).
- Congenital dermal melanocytosis all over the body, more marked on the back (Figure 2).
- Height: 68 cm; Weight: 5.3 kg (below the 3rd percentile in the WHO growth chart).
- Vital signs:
- BP: 80/60 mmHg
- PR: 68 beats per minute
- RR: 40 breaths per minute
- Temperature: 98°F
Neurological Examination
- Inability to hold the neck, sit, or roll over.
- Regression of developmental milestones.
- Normal eye movements.
Abdominal Examination
- Distended abdomen on inspection.
- Liver enlargement: 5 cm below the costal margin on the right side, total span of 10 cm (firm and non-tender).
- Spleen enlargement: 8 cm below the left costal margin.
Other Systemic Examination
- Eye movements were normal.
- Gastrointestinal, cardiovascular, and genitourinary assessments are unremarkable.
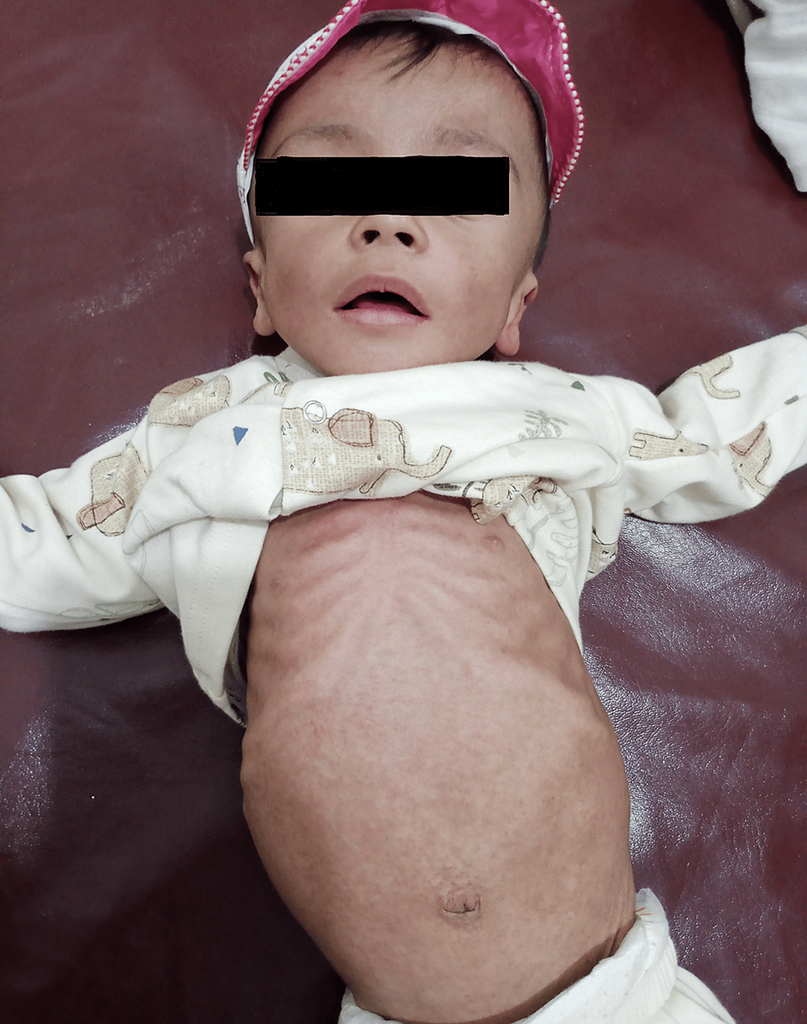
Figure 1: The photograph shows a severely wasted infant with coarse facial features and hepatosplenomegaly
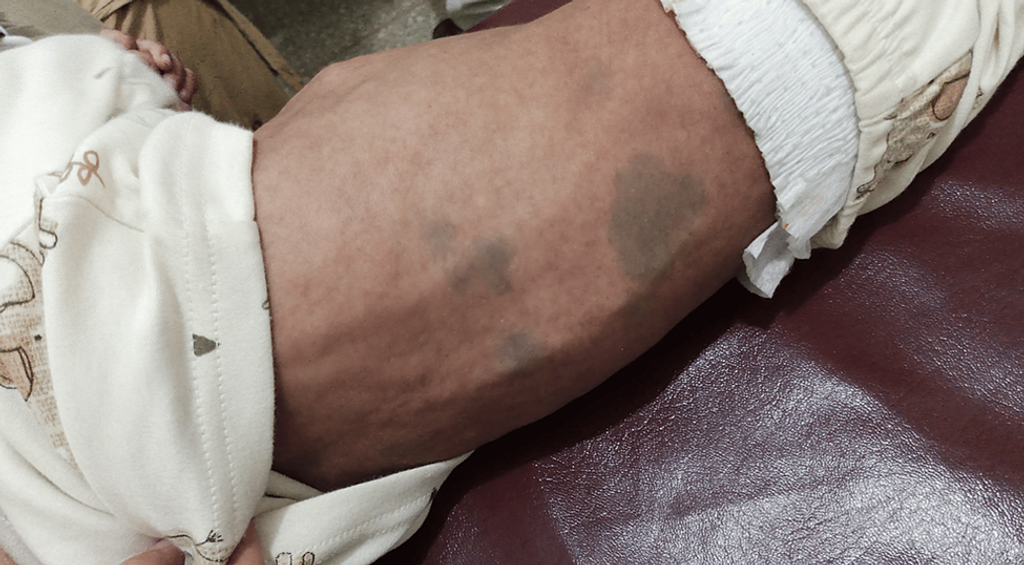
Figure 2: The photograph shows congenital dermal melanocytosis on the back
Hematological Assessment
- Complete blood count and peripheral smear findings
- Hypochromic microcytic anemia:
- Hemoglobin: 8.0 mg/dl
- Mean corpuscular volume: 59.5 fl
- Mean corpuscular hemoglobin concentration: 31.6 g/dl
- Packed cell volume: 25.2%
- Normal total leukocyte count: 9000/cm2
- Normal platelet count: 2500000/cm2
- Reticulocyte count: 2.5%
Liver Function Tests
- Liver enzymes:
- Aspartate transaminase (AST)
- Alanine transaminase (ALT)
- Alkaline phosphatase (ALP)
- Gamma-glutamyltransferase (GGT)
- Serum albumin and total protein: Normal
Metabolic and Systemic Evaluation
- Serum electrolytes, random blood sugar, serum albumin, serum cholesterol, prothrombin time, and thyroid function tests: Within normal range
Radiological Investigations
- Chest X-ray: Normal
- Ultrasound abdomen: Hepatosplenomegaly detected
Neuroimaging Findings
- MRI (magnetic resonance imaging) of the brain:
- Reduced hypointense signal in white matter bilaterally, particularly in subcortical regions
- The thinning of the corpus callosum is evident (Figure 3).
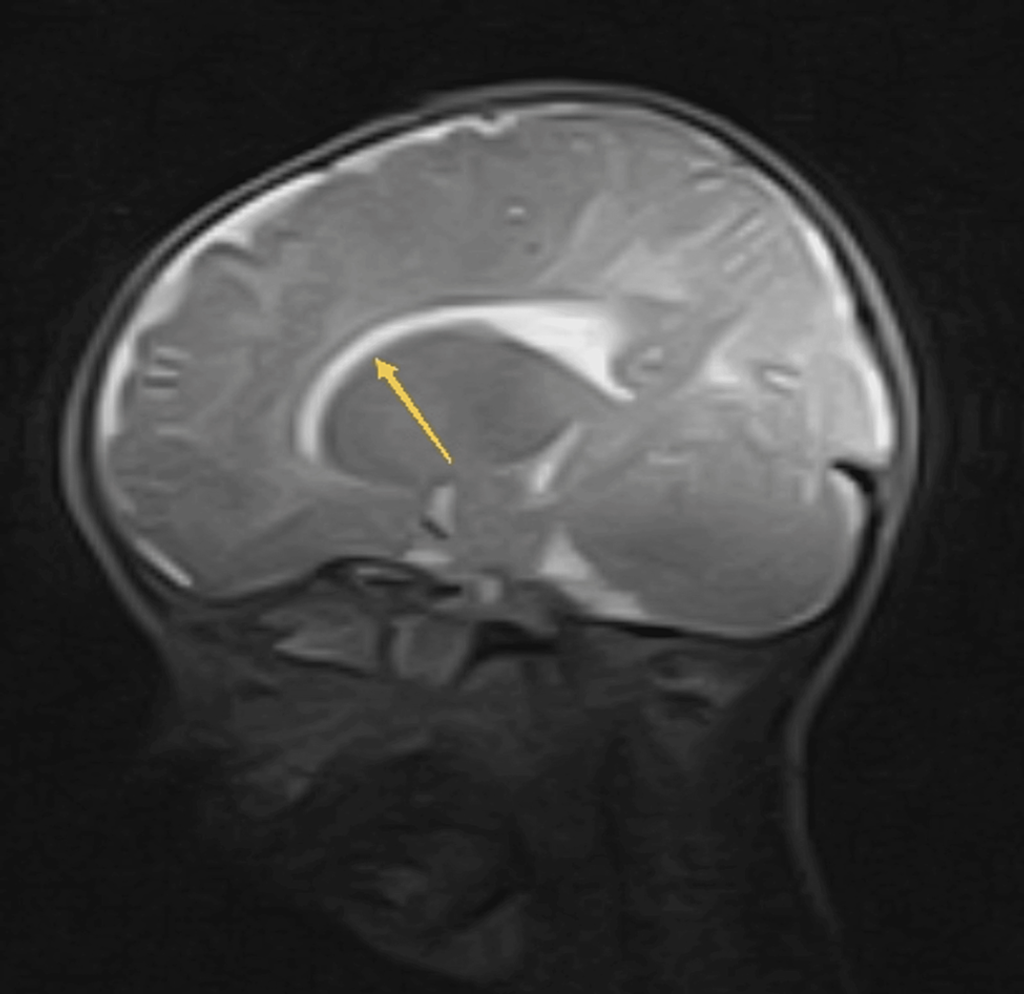
Figure 3: MRI brain T2W image shows thinning of corpus callosum (yellow arrow).
MRI: Magnetic resonance imaging; T2W image: T2-weighted image
Diagnostic Findings
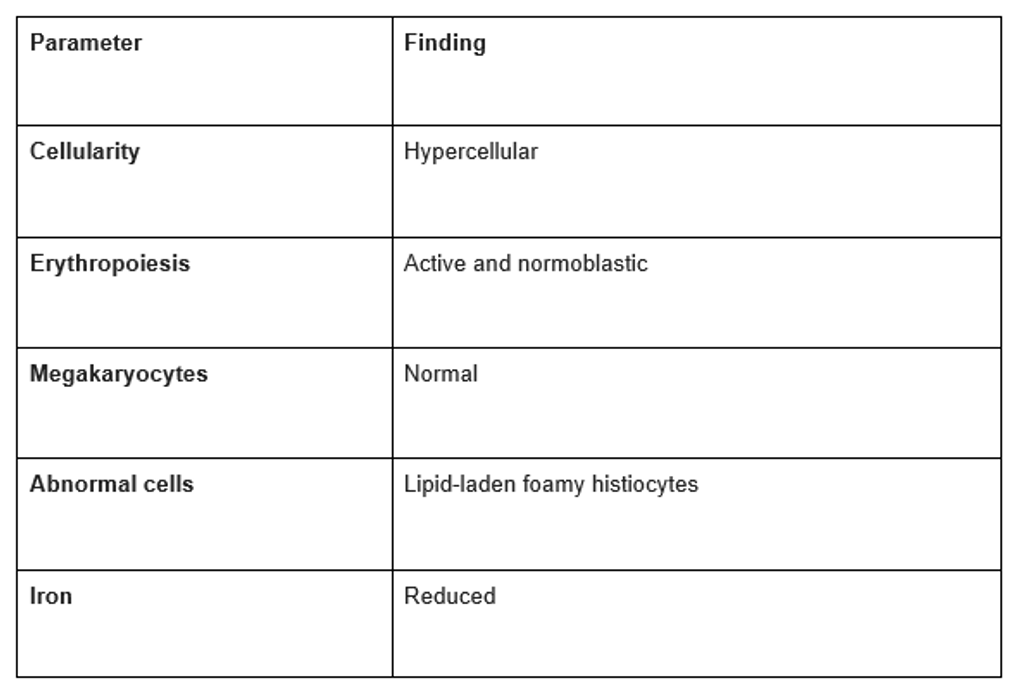
- Bone Marrow Aspiration:
- Revealed lipid-laden foamy histiocytes characteristic of Niemann-Pick disease (Figure 4).
- Normoblastic erythroid hyperplasia.
- Reduced iron levels (Table 1).
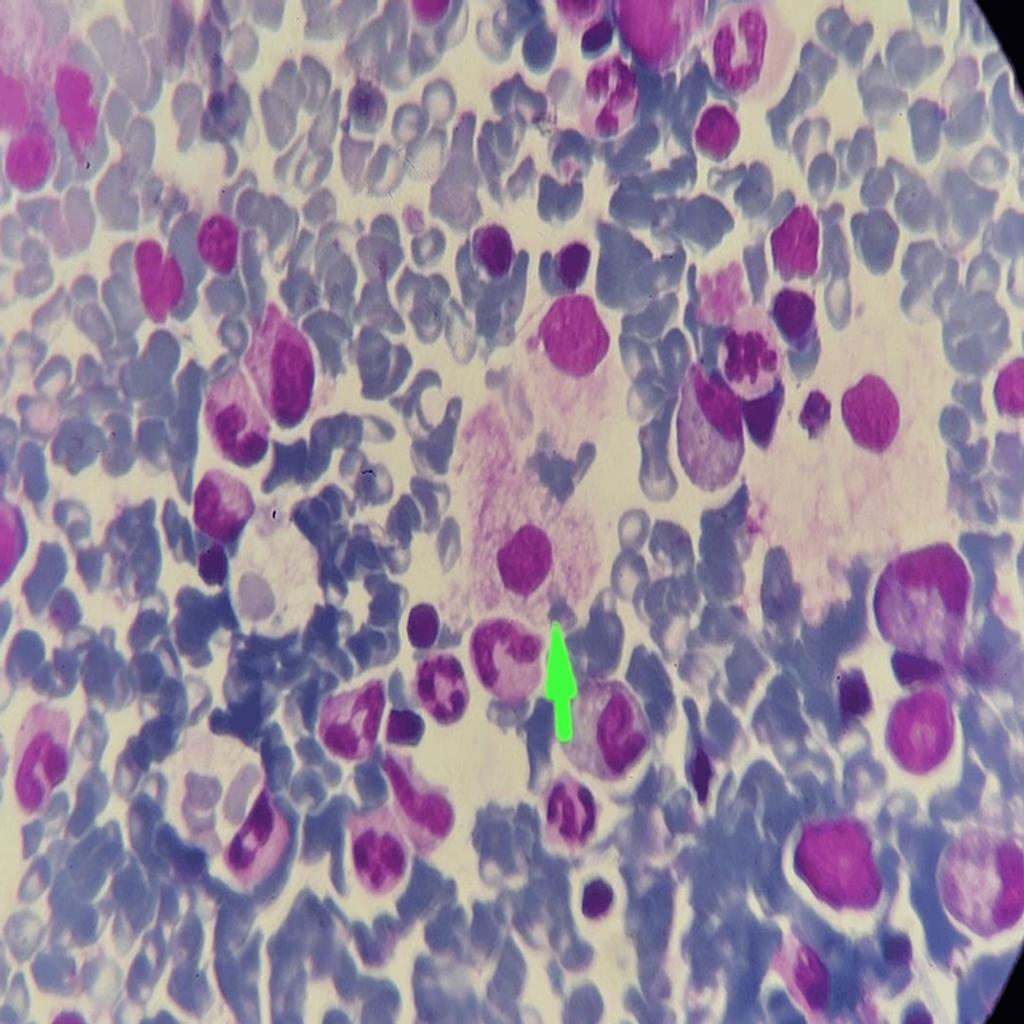
Figure 4: Bone marrow aspiration histopathological examination (100x) shows lipid-laden foamy histiocytes giving soap bubble appearance (green arrow) on Giemsa stain.
Table 1: Bone marrow aspiration report.
- Fundoscopic Examination: No macular cherry-red spot observed.
- Enzyme Activity Test: An acid sphingomyelinase enzyme activity test was advised but could not be performed due to non-availability.
- Liver Biopsy: Showed lipid-laden foamy histiocytes, confirming Niemann-Pick disease type A (NPD-A).
Diagnosis and Treatment
- Diagnosis: NPD-A diagnosis was made based on a thorough history, physical examination, and investigations
- Treatment
- No specific treatment is available for NPD-A
- Supportive care provided
- Symptomatic treatment administered
- Parents counseled on disease genetics, prognosis, and outcome
Follow-up and Current Status
- Patient’s Age: Currently 19 months old
- Follow-up: Regular follow-ups conducted
Two episodes of chest infections were reported during the follow-up period.
Discussion
Niemann-Pick disease is a group of autosomal recessive disorders marked by the accumulation of lipids, such as sphingomyelin and cholesterol, within the body. Also known as sphingomyelin-cholesterol lipidosis, this condition is caused by a deficiency in the acid sphingomyelinase enzyme. This deficiency leads to sphingomyelin and precursor lipids in lysosomes, especially in macrophages, which accumulate in organs such as the spleen, liver, brain, and lungs. This results in hepatosplenomegaly, neurological symptoms, lung diseases, and cytopenias [4,7]. There are three primary types of Niemann-Pick disease: NPD-A, NPD-B, and NPD-C. Types NPD-A and NPD-B are caused by missense mutations in the sphingomyelin phosphodiesterase 1 (SMPD1) gene on chromosome 11p15 [4,7]. NPD-C is typically due to mutations in the NPC1 gene (95% of cases) and, less commonly, the NPC2 gene (4% of cases) [6].
NPD-A, also known as the infantile neurovisceral form, is caused by extremely low activity of acid sphingomyelinase and is typically fatal by the age of three. It presents within the first few months of life with growth retardation and hepatosplenomegaly. By age one, affected children begin to show regression in developmental milestones [2,4,7]. NPD-B, which has a later onset and can affect both children and adults, does not involve neuronal tissue. Patients with NPD-B experience hepatosplenomegaly and interstitial lung disease, leading to thrombocytopenia, recurrent chest infections, and impaired bone growth. NPD-C, resulting from defective cholesterol transport, primarily affects children but can occur at any age. It leads to quadriparesis, severe lung and liver disease, supranuclear gaze palsy, dysphagia, ataxia, and dystonia.
NPD-A diagnosis is based on a thorough history, physical examination, and laboratory investigations. Clinical signs include failure to thrive, hepatosplenomegaly, and developmental delays starting at three to six months of age. Neurological symptoms, such as regression of developmental milestones and slowing of psychomotor abilities, become evident by the age of one year [8]. A macular cherry-red spot is seen in 50% of patients upon fundoscopy [4]. Laboratory tests include peripheral smears, thyroid function tests, and lipid profiles. Bone marrow aspiration histopathology can aid diagnosis by revealing lipid-laden foamy macrophages. Confirmation is achieved by measuring leukocyte acid sphingomyelinase activity and conducting genetic analysis for gene mutations [8,9].
This case report describes an 11-month-old male infant exhibiting classical features of Niemann-Pick disease type A (NPD-A), such as failure to thrive, hepatosplenomegaly, and developmental delay. Failure to thrive, frequently seen in affected infants, is due to impaired metabolism and sphingomyelin accumulation, which disrupts normal cellular functions. Hepatosplenomegaly is common in Niemann-Pick disease due to lipid storage in the liver and spleen. Developmental delay is a prominent disease feature, typically noticeable around six months of age [10]. In this case, the patient displayed delayed milestones and subsequent regression, consistent with the neurodegenerative progression of NPD-A. NPD-A is genetically based on mutations in the SMPD1 gene, leading to a deficiency in the acid sphingomyelinase enzyme. This enzyme breaks down sphingomyelin into ceramide and phosphocholine. The lack or reduced activity of acid sphingomyelinase causes sphingomyelin to accumulate in various tissues and organs, producing the disease’s characteristic features. Although the acid sphingomyelinase enzyme activity assay was not performed in this patient, the clinical presentation and the presence of lipid-rich foamy histiocytes in bone marrow and liver biopsies strongly support the diagnosis of NPD-A.
The prognosis for NPD-A is generally poor, with a life expectancy of about three years [9]. No specific treatment is available, so management focuses on supportive care to address symptoms and complications [11]. Symptomatic treatment may include nutritional support, respiratory care, and physical therapy. Parents of affected children should receive thorough counseling about the genetic nature of the disease, its expected progression, and the importance of supportive care. Early recognition and diagnosis can help provide appropriate supportive care and genetic counseling for affected individuals and their families. Since current therapies for NPD-A are purely supportive, further research is needed to explore new treatment options. Raising awareness among healthcare professionals about the clinical features and diagnostic indicators of Niemann-Pick disease can lead to earlier detection and intervention, potentially improving patient outcomes and quality of life.
Conclusions
NPD-A is a rare and fatal autosomal recessive disease that begins in the first few months of life and progressively worsens, typically leading to death by the age of three. Although no curative treatment exists for NPD-A, management focuses on supportive care to optimize the patient’s quality of life. This care involves a multidisciplinary team, including nutritionists, physiotherapists, and occupational therapists. Consanguinity is a significant modifiable risk factor for NPD-A due to its autosomal recessive inheritance. Further research is crucial to enhance our understanding of NPD-A and to develop potential therapeutic interventions. Experimental treatments like gene and enzyme replacement are being investigated and may eventually become the primary treatment options. This case report underscores the clinical features, diagnostic process, and management strategies for NPD-A, contributing to increased awareness and understanding of this severe condition among healthcare professionals.
References
- Gul F, Begum S, Rasool P, et al. (April 30, 2024) A Rare Case of Niemann-Pick Disease Type-A. Cureus 16(4): e59427. doi:10.7759/cureus.59427.
- Aghamahdi F, Nirouei M, Savad S: Niemann-Pick type A disease with new mutation: a case report. J Med Case Rep. 2022, 16:288. 10.1186/s13256-022-03486-5
- Tangde A, Pore S, Kulkarni A, Joshi A, Bindu R: Niemann-Pick disease type A-a case report. Int J Res Med Sci [Internet]. 2017, 6:366-369. 10.18203/2320-6012.ijrms20175752.
- National Institute of Neurological Disorders and Stroke (NINDS). Niemann-Pick disease. (2024). Accessed: April 30, 2024: https://www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page.
- Jeon EY, Choi KA, Koo CH, et al.: A type A Niemann-Pick disease case. J Korean Pediatr Soc. 1998, 41:275-280.
- Kundu GK, Anwar SS, Liza NAS, et al.: Niemann Pick disease: a rare lysosomal storage disease. Bangabandhu Sheikh Mujib Med Univ J. 2023, 15:141-144. 10.3329/bsmmuj. v15i2.60871.
- Vanier MT: Niemann-Pick disease type C. Orphanet J Rare Dis. 2010, 5:16. 10.1186/1750-1172-5-16 Vélez Pinos PJ, Saavedra Palacios MS, Colina Arteaga PA, Arevalo Cordova TD: Niemann-Pick disease: a case report and literature review. Cureus. 2023, 15: e33534. 10.7759/cureus.33534.
- Qureshi K, Abdulmajeed ZG, Saleem S, Tariq J, Iqbal M: Niemann-Pick disease type A: a rare disease with a fatal outcome. Cureus. 2022, 14: e21955. 10.7759/cureus.21955.
- Rajkumar V, Dumpa V: Lysosomal storage disease. In: StatPearls [Internet]. StatPearls Publishing, Treasure Island (FL); 2024.
- Wasserstein M, Dionisi-Vici C, Giugliani R, et al.: Recommendations for clinical monitoring patients with acid sphingomyelinase deficiency (ASMD). Mol Genet Metab. 2019, 126:98-105. 10.1016/j.ymgme.2018.11.014.
- Shubhankar M, Sunil KA, Bikash RP, Shantanu KM: Niemann Pick disease type A in an Infant: a case report. Sch Acad J Biosci. 2014, 2:728-730.
About Docquity
If you need more confidence and insights to boost careers in healthcare, expanding the network to other healthcare professionals to practice peer-to-peer learning might be the answer. One way to do it is by joining a social platform for healthcare professionals, such as Docquity.
Docquity is an AI-based state-of-the-art private & secure continual learning network of verified doctors, bringing you real-time knowledge from thousands of doctors worldwide. Today, Docquity has over 400,000 doctors spread across six countries in Asia. Meet experts and trusted peers across Asia where you can safely discuss clinical cases, get up-to-date insights from webinars and research journals, and earn CME/CPD credits through certified courses from Docquity Academy. All with the ease of a mobile app available on Android & iOS platforms!