This study presents a unique case of Klinefelter syndrome, a chromosomal disorder characterized by a typical karyotype of 47, XXY, accompanied by a rare mosaic form of 47, XXY, and 48, XXXY, associated with various neurological manifestations. The case involves the coexistence of spastic paraplegia and peripheral motor neuropathy, marking it as the first reported instance of such a combination in Klinefelter syndrome. Clinical examination revealed spasticity, hyperreflexia, pathological reflexes, ankle clonus, and muscle weakness affecting all extremities. Findings from motor nerve conduction study and magnetic motor evoked potential examinations indicated the presence of motor axonal neuropathy and corticospinal tract disorders.
This case highlights the potential for Klinefelter syndrome to manifest with both upper and lower motor neuron degeneration.
Introduction on Klinefelter Syndrome
Klinefelter syndrome, a chromosomal disorder characterized by the addition of an extra X-chromosome to the normal karyotype 46, XY, typically presents with a karyotype of 47, XXY, although variations including 48, XXXY; 48, XXYY; 49, XXXXY, and mosaic forms (46, XY/47, XXY and 48, XXXY/49, XXXXY) have been documented. Common clinical features include small testes, gynecomastia, hypergonadotropic hypogonadism, and neurological symptoms such as mental retardation and intellectual disability. However, instances of spastic paraplegia and peripheral neuropathy are infrequently reported in patients with Klinefelter syndrome.
This report presents a rare case of Klinefelter syndrome (mosaic form 47, XXY/48, XXXY) accompanied by both spastic paraplegia and peripheral neuropathy.
Case Presentation
1. Patient Background
- A 10-year-old boy was diagnosed with mental retardation without familial neurological history.
- Resided in a handicapped facility for 45 years without motor disabilities.
- Developed mild muscle weakness at 55 years in right upper and lower extremities (U/E and L/E, respectively), facing writing and walking difficulties.
- Progression: After a year, the inability to raise a right arm or stand-alone leads to a local hospital visit.
- Symptoms: Spasticity, muscle weakness in all extremities (A/E, R>L), bilateral gynecomastia.
- Subsequent admission to our hospital for further examination.
2. Physical Examination
- Height: 156 cm, Weight: 52 kg.
- Characteristics: Childish face, feminine voice, bilateral gynecomastia, micropenis (5 cm from pubis), absence of palpable left testis, sexual perversion (Figure A, B, C).
3. Neurological Examination
- Muscle weakness (R>L, distal>proximal), spasticity, hyperreflexia in A/E.
- Pathological Reflexes: Snout, Hoffmann, Trömner, Wartenberg, Babinski, Chaddock.
- Bilateral ankle clonus.
- FSIQ: Decreased to 61 (average: 90-110).
4. Laboratory Findings
- Serum Analysis: Elevated erythrocyte sedimentation rate (56/89 mm for 1/2 h).
- Pituitary Hormone Analysis: Low testosterone (0.5 ng/mL, normal range 2.0-7.5 ng/mL), high levels of FSH (63.7 mIU/mL, normal range 2.0-8.3 mIU/mL) and LH (32.9 mIU/mL, normal range 0.8-5.7 mIU/mL).
- Cerebrospinal Fluid Test: Normal findings.
5. Imaging Studies
- CT scan (Chest and Abdomen): Muscle atrophy of A/E, left cryptorchidism.
- MRI (Head): Normal pituitary with no significant changes.
- 99mTc-ECD-SPECT: Hypoperfusion in bilateral occipital lobe and cerebellum (Figure D & E).
6. Nerve Conduction Studies
- Motor Nerve Conduction Study: Low amplitude (1.4 mV, normal >5.5 mV), normal nerve conduction velocity in the right ulnar nerve.
- Sensory Nerve Conductions (Right Ulnar Nerve): Normal, but absence of F wave.
- Magnetic Motor Evoked Potential (MEP): No derivation on right scalp stimulation.
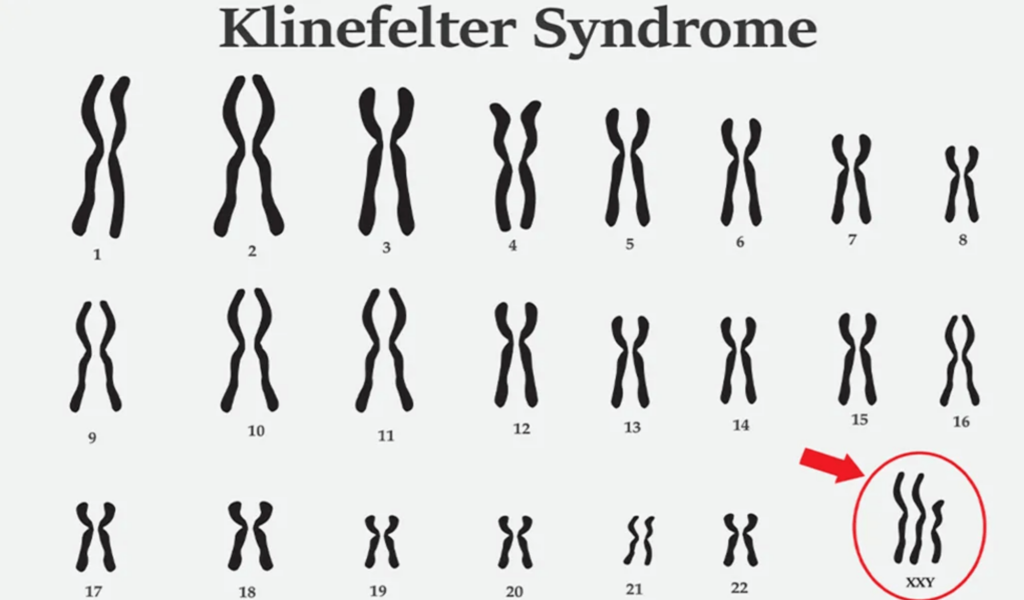
7. Chromosome Analysis
- Mosaic form of 47, XXY (2/30 cells) and 48, XXXY (20/30 cells), suggesting Klinefelter syndrome (Figure F).
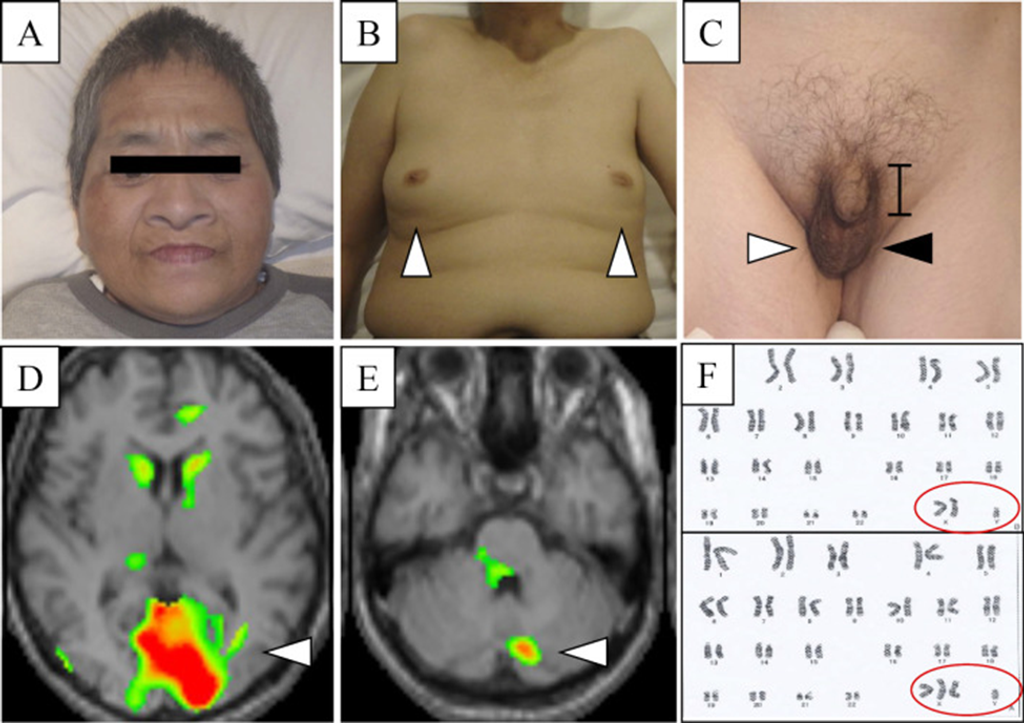
(A) The childish face of the present case. (B) Bilateral gynecomastia (arrowheads) in the present case. (C) Micropenis (bar), right normal testis (white arrowhead), and empty left scrotum (black arrowhead) in the present case. (D, E) 99mTc-ECD-SPECT showing hypoperfusion in the bilateral occipital lobe (D, arrowhead) and cerebellum (E, arrowhead). (F) A chromosome analysis showing mosaic form of 47, XXY/48, XXXY. 99mTc-ECD-SPECT: 99mTc-ethylcysteinate dimer single-photon emission computed tomography
8. Diagnosis
- The patient received a final diagnosis of Klinefelter syndrome, alongside spastic paraplegia and motor axonal neuropathy.
9. Treatment Plan
- Scheduled surgical removal of the undescended left testis from the abdominal cavity to prevent testicular cancer.
- Transfer to a local hospital for the surgical procedure and subsequent follow-up care.
Discussion
This case exhibited pyramidal signs, such as spasticity, hyperreflexia with pathological reflexes, and ankle clonus. Additionally, there was muscle weakness observed in A/E (R>L), along with mental retardation, a youthful facial appearance (Figure A), a feminine voice, bilateral gynecomastia (Figure B), and micropenis (Figure C). Hormonal analysis revealed low testosterone levels (0.5 ng/mL) and elevated FSH (63.7 mIU/mL) and LH (32.9 mIU/mL) levels. A motor nerve conduction study of the right ulnar nerve indicated motor axonal neuropathy, while MEP analysis suggested corticospinal tract disorders. Based on chromosome analysis findings, showing a mosaic form of 47, XXY (2/30 cell) and 48, XXXY (20/30 cell), the patient received a diagnosis of Klinefelter syndrome (Figure F), accompanied by both spastic paraplegia and motor axonal neuropathy.
Klinefelter syndrome is a genetic disorder characterized by additional copies of the X chromosome. These surplus X chromosomes lead to reduced testosterone production and smaller testicles, resulting in various symptoms such as increased height, gynecomastia, small testes, decreased verbal intelligence, and heightened risk of complications, including diabetes mellitus, hypothyroidism, osteoporosis, and cryptorchidism. The typical chromosomal pattern associated with Klinefelter syndrome is 47, XXY. Newborn screening research indicates an incidence rate of 1 in 650 male infants for 47, XXY. Less common chromosomal variations include 48, XXYY, 48, XXXY, and 49, XXXXY, with a rare occurrence of 1 in 50,000 male infants. Additionally, unusual mosaic patterns, as seen in the current case (47, XXY and 48, XXXY) (Figure F), have also been documented.
In this investigation, chromosome counting was conducted utilizing the Giemsa-banding (G-band) method, which employed a commercially available test kit (SRL, Tokyo, Japan). However, the limitations of the G-band method include challenges in detecting small chromosomes that are measured at less than 5 Mb. Consequently, technicians may miscount small chromosomes. Moreover, the G-band method may yield variable measurements due to reliance on technicians’ visual assessment, thereby introducing the possibility of human error. Alternative chromosome counting techniques, such as single-cell sequencing or multiplex multicolor banding, offer mechanical analysis of small chromosomes (≤500 kb). Further investigation is warranted to ascertain the most precise method for chromosome counting.
Table 1: Previous Reports of Klinefelter Syndrome Accompanied with Neuropathy or Spasticity.
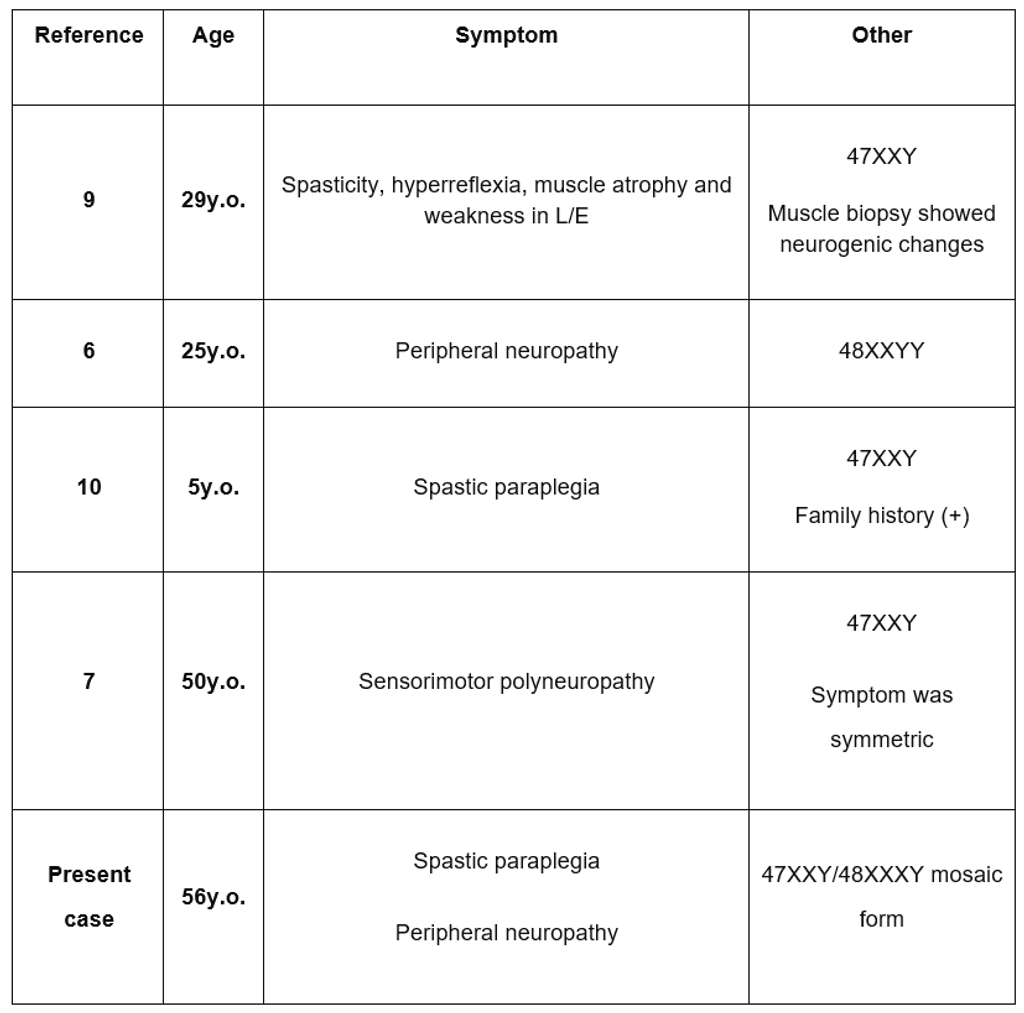
Previous literature reports have documented only two instances of Klinefelter syndrome co-occurring with neuropathy (see Table 1), likely attributed to testosterone deficiency. A prior study indicated that surplus X chromosomes disrupt testosterone production, decreasing nerve fibers containing NOS and impairing nerve function. Testosterone deficiency can also trigger conditions like diabetes mellitus and hypothyroidism, often accompanied by peripheral neuropathy. However, the current case lacked diabetes mellitus or hypothyroidism. Additionally, there have been only two reported cases of Klinefelter syndrome presenting with both spastic paraplegia and motor neuropathy. These instances imply a link between the upper and lower motor neuron degeneration and the additional X chromosome in Klinefelter syndrome.
Conclusion
In the investigation, we explored the possibility of several major hereditary spastic paraplegias (SPGs), such as SPG3A, SPG4, and SPG31. However, these typically manifest as a pure form of SPG, primarily characterized by spasticity alone. On the other hand, the current case exhibited a complex presentation of SPG, involving multiple additional symptoms alongside spasticity. Despite our thorough review, we found no hereditary SPG types that precisely aligned with the constellation of symptoms observed in our patient, which included peripheral neuropathy and mental retardation, but notably lacked cerebellar ataxia or abnormal findings on brain MRI. Thus, based on our analysis of prior cases of Klinefelter syndrome, we speculate that our case represents a novel presentation of Klinefelter syndrome accompanied by SPG and peripheral neuropathy. However, it’s important to acknowledge that we did not conduct an exome analysis in this instance, a significant limitation of our case report. Additional investigations will be necessary to exclude potential concurrent gene mutations.
In conclusion, the first documented case of Klinefelter syndrome featuring a rare mosaic chromosomal configuration of 47, XXY, and 48, XXXY, alongside manifestations of spastic paraplegia and motor axonal neuropathy. This case underscores the possibility of Klinefelter syndrome manifesting with upper and lower motor neuron degeneration.
References
- Lanfranco F, Kamischke A, Zitzmann M, Nieschlaget E. Klinefelter’s syndrome. Lancet 364: 273-283, 2004.
- Frühmesser A, Kotzot D. Chromosomal variants in Klinefelter syndrome. Sex Dev 5: 109-123, 2011.
- Tartaglia N, Ayari N, Howell S. D’Epagnier C, Zeitler P. 48, XXYY, 48, XXXY and 49, XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr 100: 851-860, 2011.
- Jesus ED, Maribel A, Eliakym A, et al. 47, XXY/48, XXXY/49, XXXXY mosaic with hydrocephaly. J Med Case Rep 1: 94, 2007.
- Bakker B, van den, Bos H, Lansdorp PM, Foijer F. How to count chromosomes in a cell: an overview of current and novel technologies. Bioessays 37: 570-577, 2015.
- Izumi S, Tsubahara A. Improvement of peripheral neuropathy by testosterone in a patient with 48, XXYY syndrome. Tokai J Exp Clin Med 25: 39-44, 2000.
- Kararizou E, Mentis AF, Gkiatas K, Davaki P. Rare association of sensorimotor polyneuropathy and Klinefelter syndrome (47, XXY). Med Princ Pract 20: 480-482, 2011.
- Traish AM, Guay AT. Are androgens critical for penile erections in humans? Examining the clinical and preclinical evidence. J Sex Med 3: 382-407, 2006.
- Matsubara S, Yoshino M, Takamori M. Benign neurogenic amyotrophy in Klinefelter’s syndrome. J Neurol Neurosurg Psychiatry 57: 640-642, 1994.
- Uzicanin S, Catibusic F, Terzic S, Zubcevic S. Familiar spastic paraplegia presenting in a boy with Klinefelter syndrome. Med Arh 61: 52-53, 2007.
About Docquity
If you need more confidence and insights to boost careers in healthcare, expanding the network to other healthcare professionals to practice peer-to-peer learning might be the answer. One way to do it is by joining a social platform for healthcare professionals, such as Docquity.
Docquity is an AI-based state-of-the-art private & secure continual learning network of verified doctors, bringing you real-time knowledge from thousands of doctors worldwide. Today, Docquity has over 400,000 doctors spread across six countries in Asia. Meet experts and trusted peers across Asia where you can safely discuss clinical cases, get up-to-date insights from webinars and research journals, and earn CME/CPD credits through certified courses from Docquity Academy. All with the ease of a mobile app available on Android & iOS platforms!