Pompe disease, a rare autosomal recessive disorder, results from a deficiency in acid alpha-glucosidase (GAA), leading to glycogen accumulation within lysosomes and resulting in skeletal, cardiac, and smooth muscle tissue lesions. The disease progresses over time, with severity varying based on the age at onset. The most severe form, known as classic infantile Pompe disease, manifests before 12 months of age. Instances of Pompe disease with onset during intrauterine development have been exceptionally rare.
Pompe Disease: Introduction
Pompe disease, or glycogen storage type II, is a rare autosomal recessive disorder. It arises from a deficiency in acid alpha-glucosidase (GAA), resulting in glycogen accumulation within lysosomes and leading to lesions in skeletal, cardiac, and smooth muscle tissues. The progression of Pompe disease varies, with severity influenced by the age at which symptoms appear. Classic infantile Pompe disease, the most severe form, typically manifests before the age of 12 months. It is characterized by rapid progression of hypertrophic cardiomyopathy, causing left ventricular outflow obstruction, alongside generalized hypotonia, delayed motor development, feeding and swallowing difficulties, and dyspnea.
While cases of Pompe disease with onset during intrauterine development are rare, they pose diagnostic challenges due to the absence of specific manifestations. This report presents a case of infantile Pompe disease with intrauterine onset, wherein clinical symptoms appeared postnatally, accompanied by significantly reduced GAA enzymatic activity and the presence of a rare homozygous mutation detected through genetic testing. Additionally, a literature review on infantile Pompe disease with intrauterine onset is provided, summarizing clinical characteristics and genotypes of reported patients. This collective information aims to facilitate early diagnosis and prompt initiation of treatment strategies.
Case Presentation
Patient Information:
- Gender: Female
- Gestation: 40 weeks and three days
- Birth Weight: 4,090 grams
Reason for Admission:
- Condition: Intrauterine myocardial hypertrophy persisting for two weeks
- Symptoms: Shortness of breath and cyanosis noted until 13 minutes after birth
Electrocardiogram Monitoring Results:
- Heart Rate: 140 beats per minute
- Respiration Rate: 60 beats per minute
- Oxygen Saturation: Below 90%
Physical Examination Findings on Admission:
- Responsiveness: Poor
- Pallor: Present
- Breathing: Irregular
- Hoover’s Sign: Positive, without lung crackles
- Heart Sounds: Faint, no murmurs detected
- Liver: Normal size
- Limb Muscle Tone: Decreased
- Edema: Present in lower limbs
Family History:
- Parents: Healthy and unrelated
Prenatal Ultrasound Findings:
- Gestation Week: 38
- Observations: Enlarged heart with significantly thickened left ventricular walls (10.4 mm interventricular septum; 10.0 mm posterior left ventricle) and mild pericardial effusion (Fig 1).
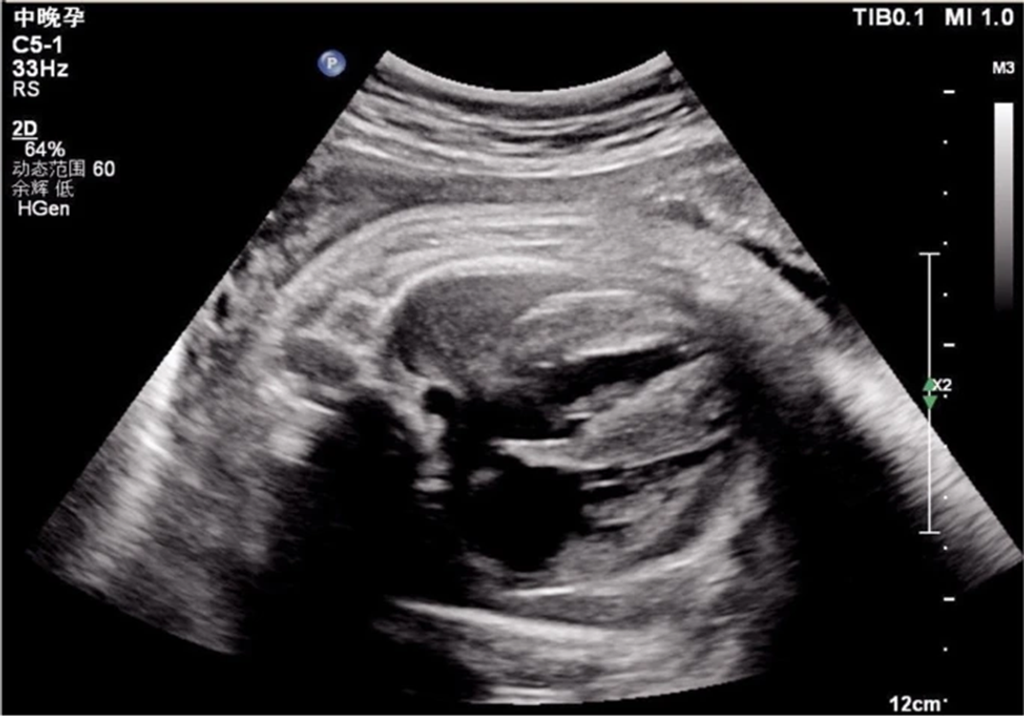
Figure 1: Echocardiogram demonstrating ventricular hypertrophy in utero
Laboratory Findings:
- B-type Natriuretic Peptide (BNP): 1,777.6 pg/mL
- Creatine Kinase (CK): 3,594 ng/mL
- Myoglobin: >1,200.00 ng/mL
Diagnostic Imaging Results:
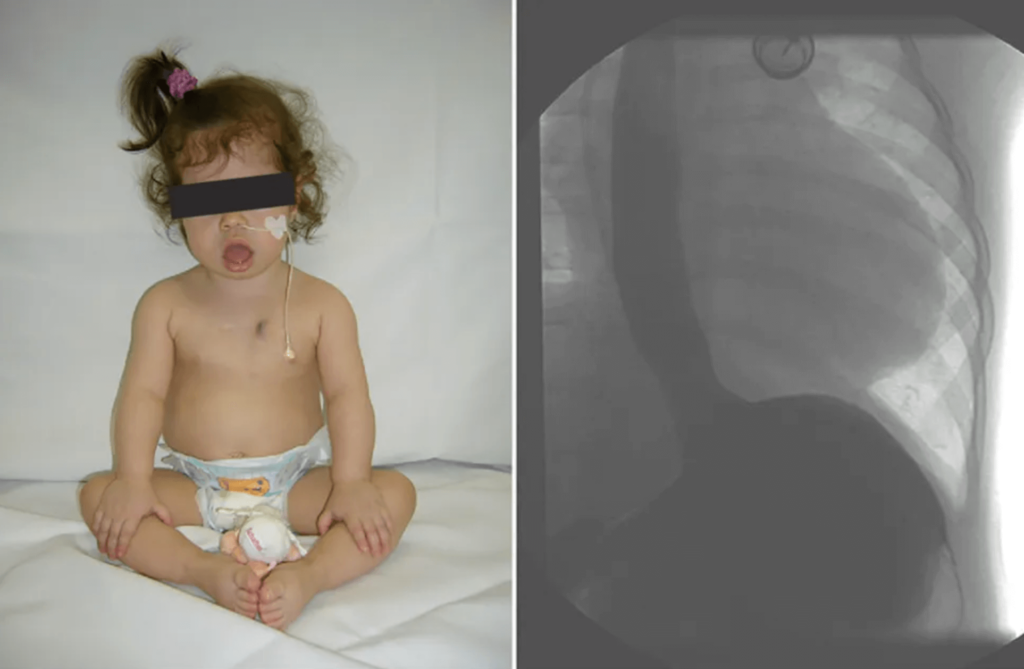
- Chest X-ray revealed a significantly increased cardiothoracic proportion (0.8) (Fig 2).
- Electrocardiogram (ECG): Showed a shortened PR interval, abnormal Q wave, and ST-T changes (Fig 3).
- Echocardiography: Indicated heart enlargement with significant left ventricular wall thickening (interventricular septum, 9 mm) (Fig 4).
Treatment Plan:
- Interventions: Non-invasive ventilator-assisted breathing, nasal feeding, metoprolol treatment to inhibit myocardial remodeling
Additional Diagnostic Tests:
- Tandem Mass Spectrometry: GAA enzymatic activity measured at 0.31 µmol/L/h
- Genetic Test: Detected missense mutation c.1844G > T (p. Gly615Val)
Diagnosis:
- Condition: Infantile Pompe disease (Fig 5).
Treatment Decision:
- Enzyme Replacement Therapy (ERT): Declined by parents due to financial constraints and perceived poor prognosis.
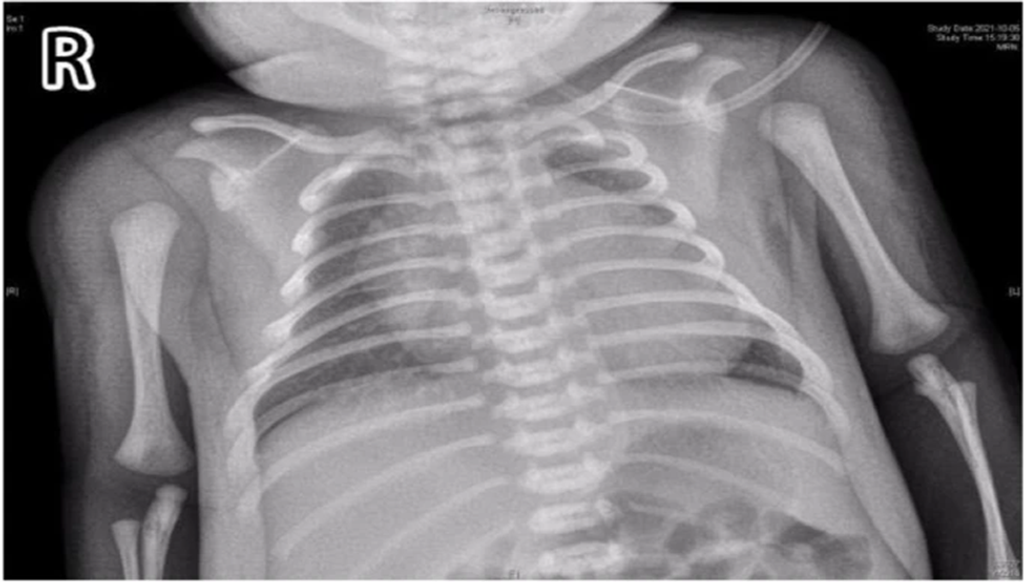
Figure 2: Chest films showing cardiomegaly with a cariothoracic ratio of approximately 80%
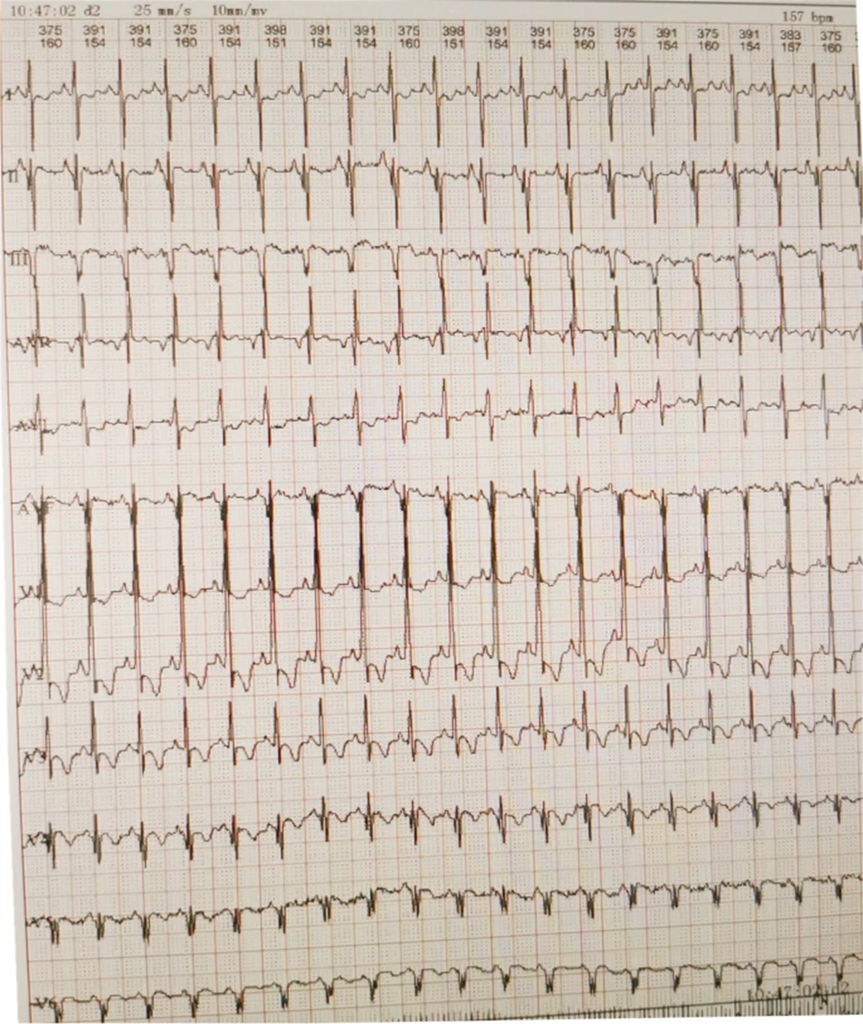
Figure 3: Electrocardiography revealing a short PR interval, high QRS voltage, ST-T changes, and a prominent Q wave
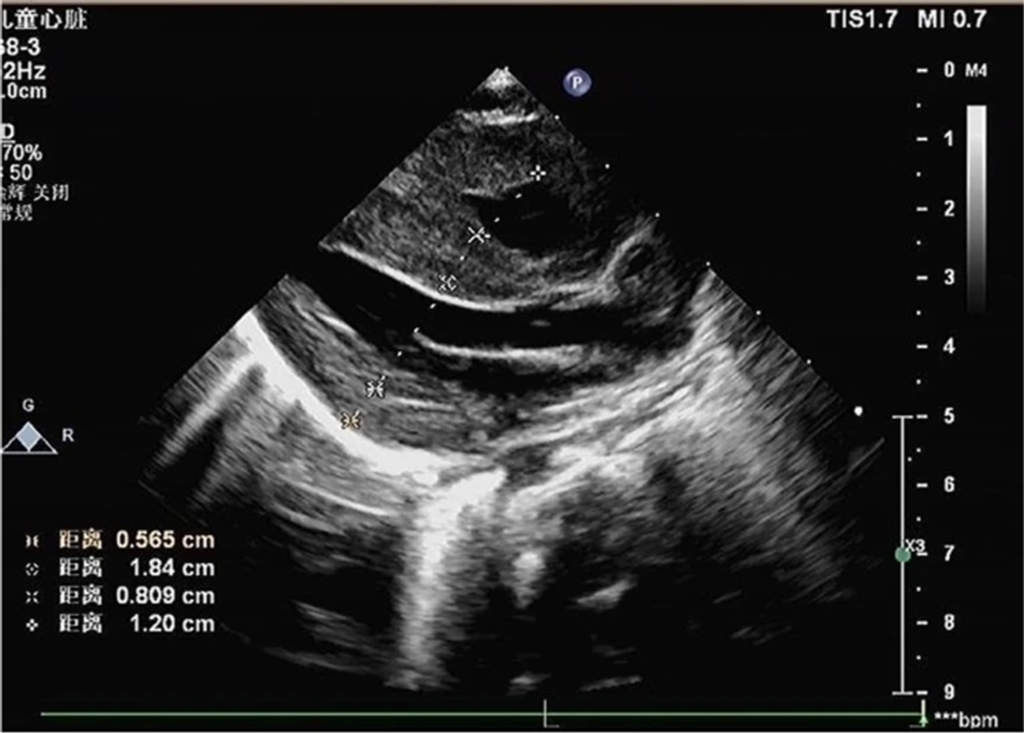
Figure 4: Echocardiogram demonstrating ventricular hypertrophy postnatally
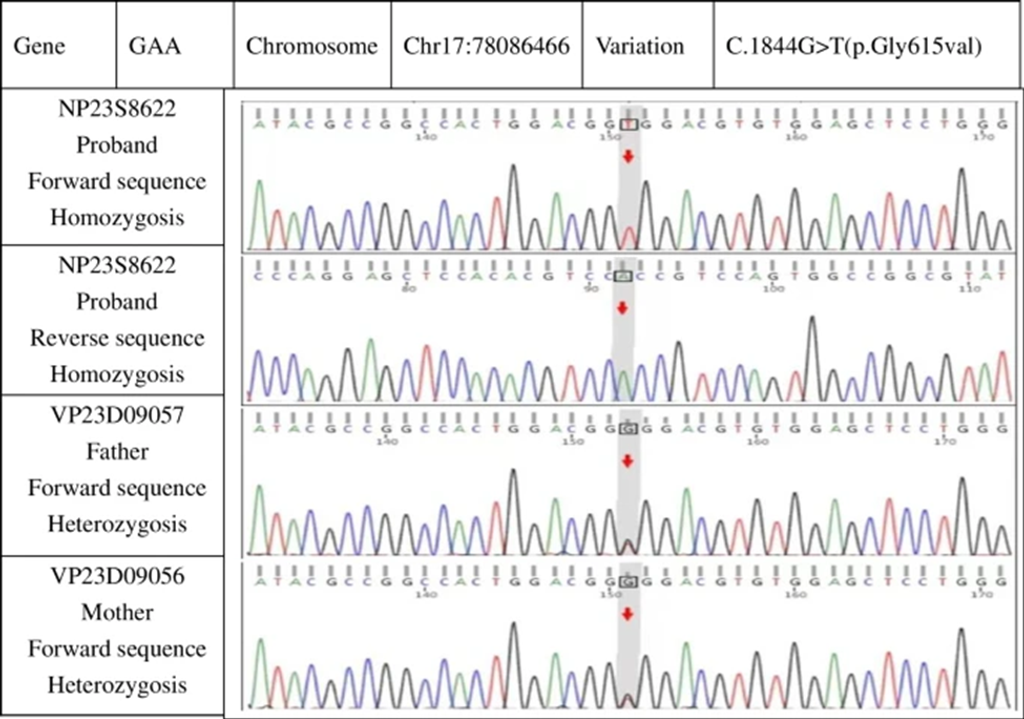
Figure 5: Genetic studies revealing a point mutation on exon 13 with Gly615Val and both parents carrying the recessive gene
Patient Information at Six Months
- Weight and Length:
- Weight: 7.5 kg
- Length: 70 cm
- Feeding and Breathing:
- Able to suck milk independently
- No breathing difficulties
- Crying and Muscle Tone:
- Hoarse and weak crying
- Significant reduction in muscle tension throughout the body
- Inability to raise head or turn over
Medical Test Results
- Blood Test:
- Creatine Kinase (CK) Level: 863.4 IU/L
- B-type Natriuretic Peptide (BNP) Level: 2,509.3 pg/mL
- Imaging and Ultrasound:
- Chest X-ray: Left lung consolidation (Fig. 6).
- Chest Ultrasound: Left thoracic solid medium echo consistent with lung tissue consolidation (Fig. 7).
- Echocardiography:
- Thickened ventricular septum and left and right ventricular free walls: 1.2 cm, 1.0 cm, and 0.65 cm, respectively.
- Increased and thickened left intraventricular and papillary muscle trabeculae
- Ejection Fraction: 57%
Outcome
- Cause of Death: Cardiopulmonary failure at seven months of age
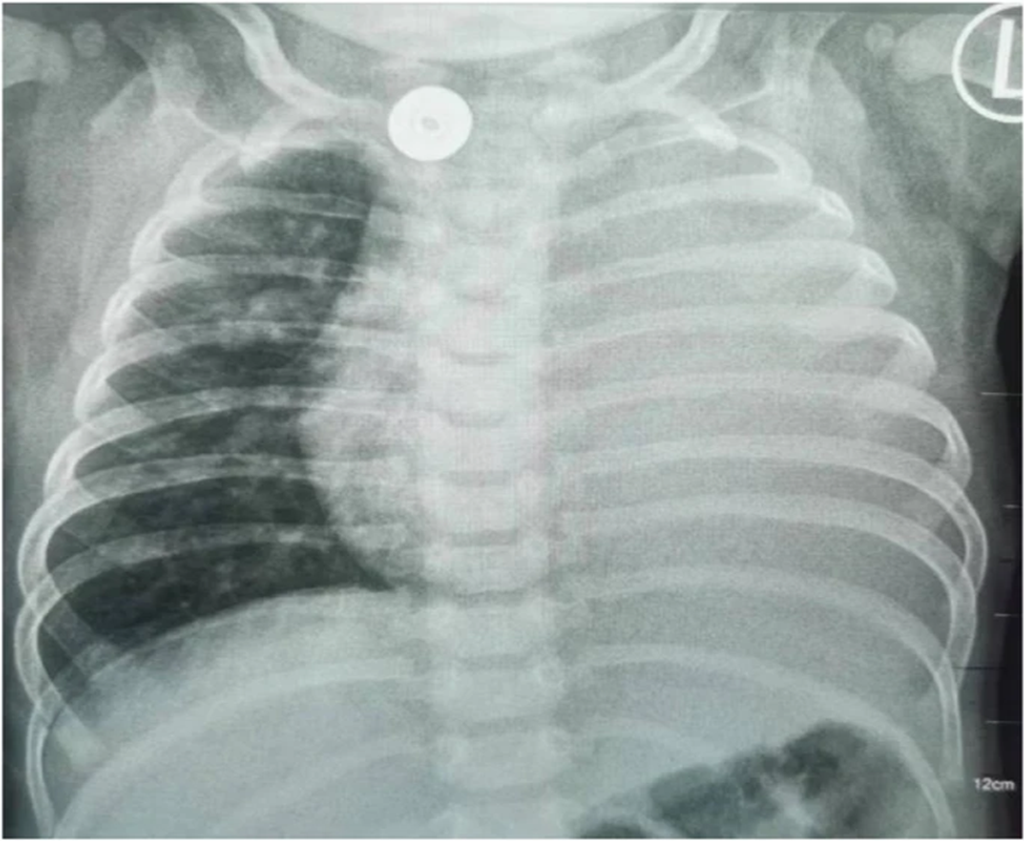
Figure 6: Patient showing left lung consolidation at six months of age
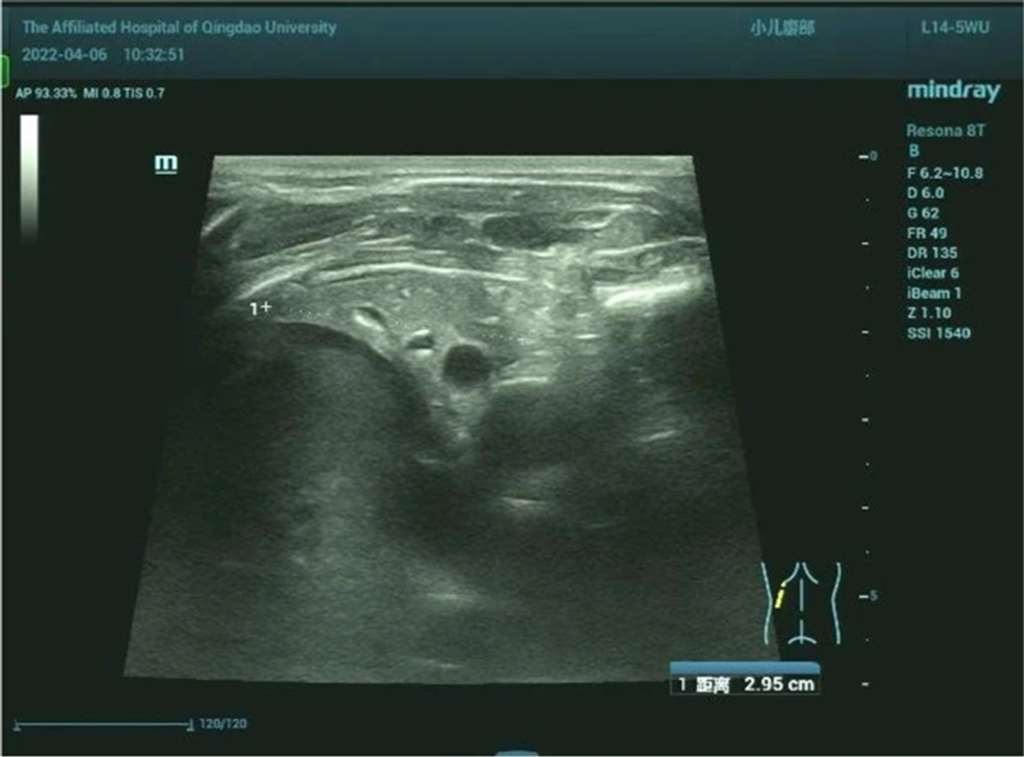
Figure 7: Left lung showing the same consolidation density as the liver
Discussion & Conclusions
Classic infantile Pompe disease typically has a median onset age of 2.4 months (range: 0.0–12.0 months) but can present clinical symptoms at any age [7]. Intrauterine onset of Pompe disease is extremely rare; including the present case, only seven such cases have been reported. Despite the term “infantile,” the disease can begin before birth. Pompe disease has various manifestations, with infantile cardiomyopathy mainly presenting as left ventricular myocardial hypertrophy [8]. All seven neonates showed myocardial changes on intrauterine ultrasound, with variations including hypertrophic cardiomyopathy, dilated cardiomyopathy, and myocardial mass. Postnatally, all cases progressed to myocardial hypertrophy, with levels of myocardial enzymes and BNP increasing to varying degrees.
Neonatal hypertrophic cardiomyopathy and hypotonia must be differentiated from other conditions such as glycogen storage type IIB (Danon disease), specific fatty acid oxidation disorders, and mitochondrial respiratory chain dysfunction [9,10,11]. These conditions have similar clinical symptoms and are primarily diagnosed through genetic testing.
All patients had GAA-related gene mutations [12]. Despite the intrauterine onset, prenatal diagnosis via GAA mutation analysis from amniocentesis was achieved in only one patient (case #5) due to positive family history [13]. Chien et al. suggested that prognosis improves significantly when treatment begins within the first few days of life, highlighting the importance of early diagnosis. Prenatal diagnosis can be based on detecting GAA deficiency in amniotic fluid cultures or identifying GAA mutations. Pompe disease is inherited recessively, and genetic diagnosis using chorionic villus samples is feasible only if the parents are known carriers. Maternal tissue contamination can lead to false-positive results in biochemical or molecular methods; thus, targeted mutational analysis is necessary to confirm prenatal diagnosis [14,15]. In this case, prenatal diagnosis was not possible due to the unknown family history.
Enzyme replacement therapy (ERT) is the cornerstone of managing Pompe disease [16]. When started early, ERT can enhance left ventricular function and yield better outcomes [17]. Therefore, newborn screening for Pompe disease is crucial [18].
The team reviewed seven patients, five receiving enzyme replacement therapy (ERT). The onset of treatment ranged from two hours postnatally (case #3) to two months (case #4). Two patients (cases #6 and #7) did not receive treatment. In all patients treated with ERT, myocardial thickness returned to normal; however, one patient (case #2) died from respiratory failure due to infection.
Our patient had homozygous gene mutations at the same site and presented with clinical symptoms postnatally. After symptomatic treatment, vital signs stabilized briefly, but the patient experienced a significant developmental delay. At six months, the patient developed lung consolidation, worsening myocardial hypertrophy, decreased cardiac function, and severe hypotonia. Respiratory failure in Pompe disease often results from extensive pathological changes in the muscles and nerves of the respiratory system due to glycogen accumulation [19]. However, no reported cases of Pompe disease have shown homogeneous solid changes in the lungs. A knockout mouse model of enzyme-deficient Pompe disease demonstrated glycogen storage in nearly all tissue and cell types [20]. Another mouse model showed lysosomal glycogen accumulation in tracheal and bronchial smooth muscles [21]. Thus, lung consolidation in patients with Pompe disease is likely related to glycogen accumulation and deposition.
Although ERT improved clinical symptoms in some patients, it has several limitations, including the risk of immunogenicity-related complications, the inability to penetrate central nervous system tissue, and the necessity for lifelong therapy. Additionally, no trials have compared the effectiveness or safety of ERT with other interventions or placebos until 2016 [22]. Gene therapy appears promising for potentially preventing, halting, and reversing Pompe disease [19]. However, only animal experiments and small clinical studies have been conducted, limiting the applicability of gene therapy [1]. None of the patients we reviewed received gene therapy.
Infantile Pompe disease is a rare hereditary condition that can occur in utero, with initial signs often being cardiac changes. Therefore, enzyme assays and genetic testing should be performed promptly to diagnose and treat fetuses with intrauterine cardiac disease early.
This study has several limitations. First, the number of cases was small. Second, follow-up for some patients had not been completed at the time of reporting.
References
- Meena NK, Raben N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules. 2020;10(9):1339. Published 2020 Sep 18. doi: https://doi.org/10.3390/biom10091339
- Llerena Junior JC, Nascimento OJ, Oliveira AS, et al. Guidelines for diagnosing, treating, and clinically monitoring patients with juvenile and adult Pompe disease. Arq Neuropsiquiatr. 2016;74(2):166–76. https://doi.org/10.1590/0004-282X20150194.
- Hamdan MA, El-Zoabi BA, Begam MA, Mirghani HM, Almalik MH. Antenatal diagnosis of Pompe disease by fetal echocardiography impact on outcome after early initiation of enzyme replacement therapy. J Inherit Metab Dis. 2010,33 Suppl 3S333-S339. https://doi.org/10.1007/s10545-010-9179-2.
- Tsai AC, Hung YW, Harding C, Koeller DM, Wang J, Wong LC. Next-generation deep sequencing corrects diagnostic pitfalls of traditional molecular approach in a patient with prenatal onset of Pompe disease. Am J Med Genet A. 2017;173(9):2500–4. https://doi.org/10.1002/ajmg.a.38333.
- Swarr DT, Kaufman B, Fogel MA, Finkel R, Ganesh J. Unusual cardiac “masses“in a newborn with infantile Pompe disease. JIMD Rep. 2012; 5:17–20. https://doi.org/10.1007/8904_2011_85.
- Gupta P, Shayota BJ, Desai AK, et al. A Race Against Time-Changing the Natural History of CRIM Negative Infantile Pompe Disease. Front Immunol. 2020; 11:1929. Published 2020 Sep 4. https://doi.org/10.3389/fimmu.2020.01929.
- Reuser AJJ, van der Ploeg AT, Chien YH, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease Data from the Pompe Registry. Hum Mutat. 2019;40(11):2146–64. https://doi.org/10.1002/humu.23878.
- Van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet. 2008;372(9646):1342–53. https://doi.org/10.1016/S0140-6736(08)61555-X.
- Gu J, Geng M, Qi M, Wang L, Zhang Y, Gao J. The role of lysosomal membrane proteins in glucose and lipid metabolism. FASEB J. 2021;35(10): e21848. https://doi.org/10.1096/fj.202002602R.
- Marsden D, Bedrosian CL, Vockley J. Impact of newborn screening on the reported incidence and clinical outcomes associated with medium- and long-chain fatty acid oxidation disorders. Genet Med. 2021;23(5):816–29. https://doi.org/10.1038/s41436-020-01070-0.
- Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17(9):689–701. https://doi.org/10.1038/gim.2014.177.
- Chien YH, Hwu WL, Lee NC. Pompe disease: early diagnosis and early treatment make a difference. Pediatr Neonatol. 2013;54(4):219–27. https://doi.org/10.1016/j.pedneo.2013.03.009.
- Bembi B, Cerini E, Danesino C, et al. Diagnosis of glycogenosis type II. Neurology. 2008;71(23 Suppl 2):S4–11. https://doi.org/10.1212/WNL.0b013e31818da91e.
- Fowler DJ, Anderson G, Vellodi A, Malone M, Sebire NJ. Electron microscopy of chorionic villus samples for prenatal diagnosis of lysosomal storage disorders. Ultrastruct Pathol. 2007;31(1):15–21. https://doi.org/10.1080/01913120601169469.
- van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24(6):768–e31. https://doi.org/10.1111/ene.13285.
- Davison JE. Advances in diagnosis and management of Pompe disease. J Mother Child. 2020;24(2):3–8. https://doi.org/10.34763/jmotherandchild.20202402si.2001.000002 (Published 2020 Oct 2).
- Yang CF, Yang CC, Liao HC, et al. Very early treatment for infantile-onset Pompe disease contributes to better outcomes. J Pediatr. 2016; 169:174–80.e1. https://doi.org/10.1016/j.jpeds.2015.10.078.
- Chien YH, Chiang SC, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008;122(1):e39–45. https://doi.org/10.1542/peds.2007-2222.
- Fusco AF, McCall AL, Dhindsa JS, et al. The Respiratory Phenotype of Pompe Disease Mouse Models. Int J Mol Sci. 2020;21(6):2256. https://doi.org/10.3390/ijms21062256 (Published 2020 Mar 24).
- van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet. 2008;372(9646):1342–53. https://doi.org/10.1016/S0140-6736(08)61555-XGaa.
- Keeler AM, Liu D, Zieger M, et al. Airway smooth muscle dysfunction in Pompe (Gaa-/-) mice. Am J Physiol Lung Cell Mol Physiol. 2017;312(6): L873–81. https://doi.org/10.1152/ajplung.00568.2016.
- Chen M, Zhang L, Quan S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst Rev. 2017;11(11): CD011539. https://doi.org/10.1002/14651858.CD011539.pub2 (Published 2017 Nov 20).
About Docquity
If you need more confidence and insights to boost careers in healthcare, expanding the network to other healthcare professionals to practice peer-to-peer learning might be the answer. One way to do it is by joining a social platform for healthcare professionals, such as Docquity.
Docquity is an AI-based state-of-the-art private & secure continual learning network of verified doctors, bringing you real-time knowledge from thousands of doctors worldwide. Today, Docquity has over 400,000 doctors spread across six countries in Asia. Meet experts and trusted peers across Asia where you can safely discuss clinical cases, get up-to-date insights from webinars and research journals, and earn CME/CPD credits through certified courses from Docquity Academy. All with the ease of a mobile app available on Android & iOS platforms!