Congenital adrenal hyperplasia (CAH) constitutes a group of genetic disorders primarily originating from mutations in the CYP21A2 gene, responsible for encoding the adrenal steroid 21-hydroxylase enzyme (P450c21). This genetic anomaly results in impaired adrenal steroidogenesis and is inherited as an autosomal recessive trait [1]. The mutation in the 21-hydroxylase enzyme leads to diminished cortisol production, triggering an elevation in corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH), ultimately causing adrenal cortex hyperplasia [2]. Consequently, an excess of adrenal androgens is produced. Clinically, CAH manifests in two forms: “classic” and “nonclassic” (NCCAH) [3], with the distinction dependent on the severity of the deficiency’s clinical expression [4]. NCCAH typically exhibits no symptoms before the age of five and is commonly diagnosed during puberty, particularly in individuals seeking fertility treatment. Notably, NCCAH shares significant phenotypic similarities with polycystic ovarian syndrome (PCOS), often leading to misdiagnosis [1].
In the context of assisted reproductive therapy for NCCAH patients, elevated progesterone concentrations during the follicular phase and anovulatory cycles associated with excessive androgens may impact tubal motility, cervical thickness, and endometrial receptivity [5]. For adult women with NCCAH not requiring fertility treatment, primary methods to manage hyperandrogenism involve oral contraceptives and anti-androgen therapy. However, in cases where these treatments are intolerable, glucocorticoid therapy becomes the preferred approach for addressing hyperandrogenic symptoms and menstrual cycle management [3,6].
The incidence of classical CAH is estimated at 1:10,000 – 1:20,000 live births, while NCCAH occurs in approximately 1:1,000 live births [7, 8]. The overall prevalence of NCCAH among women exhibiting androgen excess symptoms is around 4% [9], with variations based on racial differences placing the prevalence of 21OHD NCCAH between 1:1,000 and 1:2,000 [10].
Genetic testing is a crucial diagnostic tool for NCCAH, particularly when biochemical results are ambiguous or genetic counseling is necessary before pregnancy to identify CYP21A2 genotyping and heterozygous carriers. Although over 200 mutations of the CYP21A2 gene have been reported in various studies [11], current efforts still focus on understanding the clinical phenotype of compound CYP21A2 pathogenic mutations and their interactions.
This study not only detected a new CYP21A2 mutation through multiple genetic tests (gene sequencing, MLPA, CNVs, and SNP) but also explored the correlation between clinical manifestations and gene phenotypes associated with this novel mutation. This early misdiagnosis case offers valuable insights into NCCAH from both genetic and clinical perspectives, paving the way for future clinical and scientific research directions in the field of CAH.
Methodology [1]
Clinical Presentation
Initially misdiagnosed in prior medical facilities, the patient sought assistance in other specialized center due to Polycystic Ovary Syndrome (PCOS) and infertility issues. Despite undergoing several cycles of ovulation induction treatment, where the follicles exhibited satisfactory development, the attempts failed to result in pregnancy. Consequently, the patient needed in vitro fertilization (IVF) treatment. During the Controlled Ovarian Stimulation (COS) phase, the follicles demonstrated optimal development, increasing E2 levels; however, the progesterone levels remained abnormally elevated at 3.29-5.98 ng/ml. Oocyte Pick Up (OPU) yielded 16 eggs, from which nine blastocysts were successfully frozen. Notably, the persistent abnormal elevation of progesterone during COS distinguished the patient’s hormone profile from that of the typical PCOS population. This prompted a recommendation for genetic testing, revealing the presence of nonclassical Congenital Adrenal Hyperplasia (NCCAH) in the patient (proband). To identify the origin of the pathogenic gene and provide guidance for pregnancy, a comprehensive genetic analysis was subsequently conducted on other family members, including embryos of potential Offspring.
Genetic Testing Procedures
Sanger sequencing was employed to measure the mutation sites in the CYP21A2 gene while detecting the CYP21A1P/A2 fusion gene was carried out using Multiplex ligation-dependent probe amplification (MLPA) within the family. Preimplantation Genetic Testing for Monogenetic Disease (PGTM) was also used to assess trophoblast cells in the female proband’s embryos. This involved the utilization of Copy-Number Variations of a Single Human Cell and high-throughput sequencing. The identification of mutation sites in the CYP21A2 gene and the origin of the CYP21A1P/A2 fusion gene in each embryo was accomplished through a combination of Sanger sequencing, whole-genome sequencing (WGS), and single nucleotide polymorphism (SNP) analysis.
Results
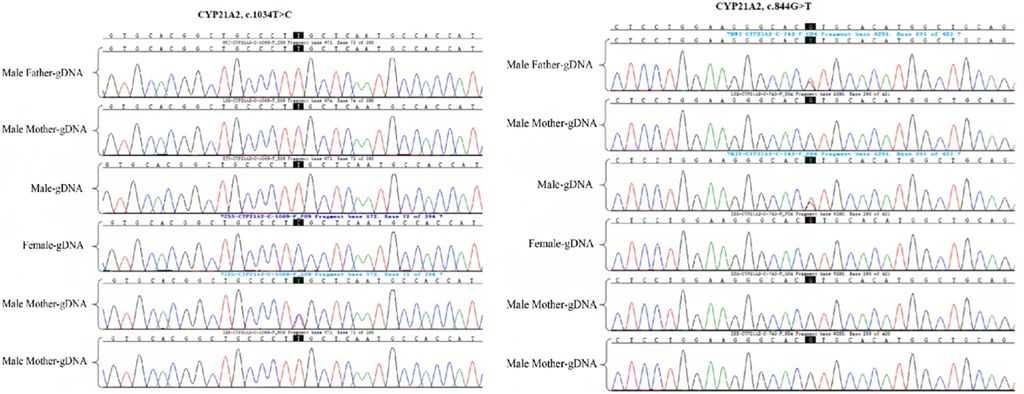
Figure 1: Detection of CYP21A2 gene mutation site in the family by Sanger sequencing. The female proband and her father had a CYP21A2 mutation gene c.1034T > C (p. L354S); moreover, the husband of the proband and the male father carried the CYP21A2 gene c.844G>T.

Figure 2: Detection of CYP21A2 fusion gene in the family by MLPA, which presented the CYP21A2 mutation gene site. The female proband and her mother carry CYP21A2, CYP21A1P/A2 fusion gene (CH-8), and MLPA showed exon1-7 deletion.
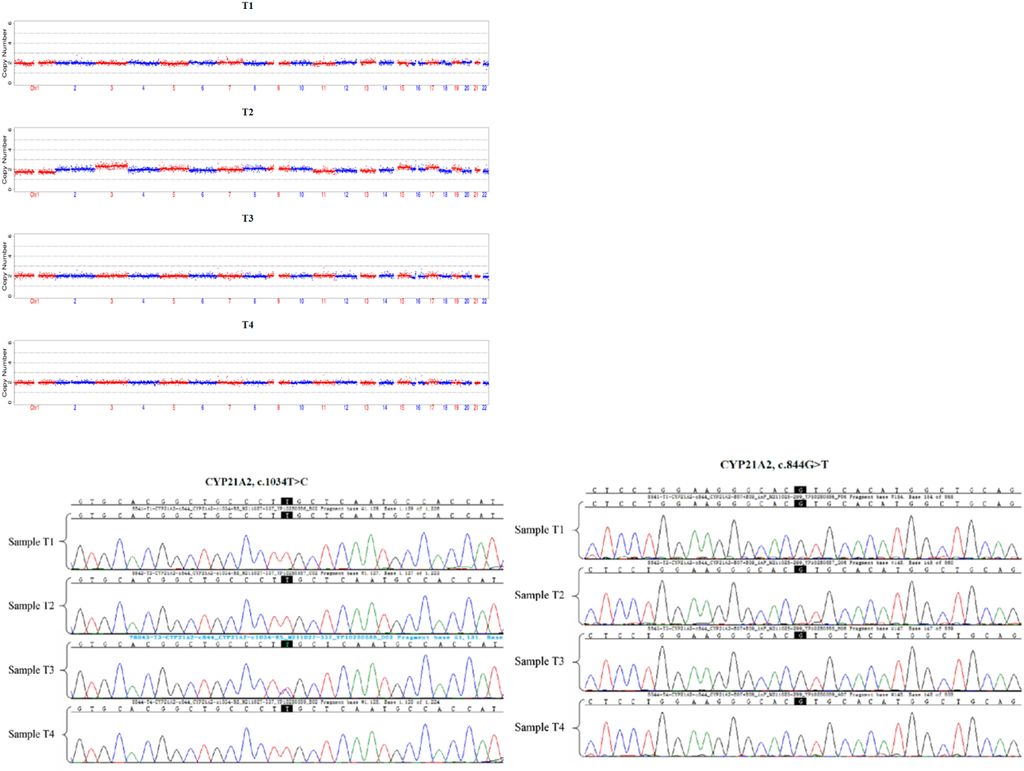
Figure 3: Detection of trophoblast cells of each embryo by CNVs and high-throughput sequencing, and CYP21A2 mutation gene site of each embryo by Sanger sequencing. The embryo T3 carried c.1034T > C heterozygous mutation of maternal origin.
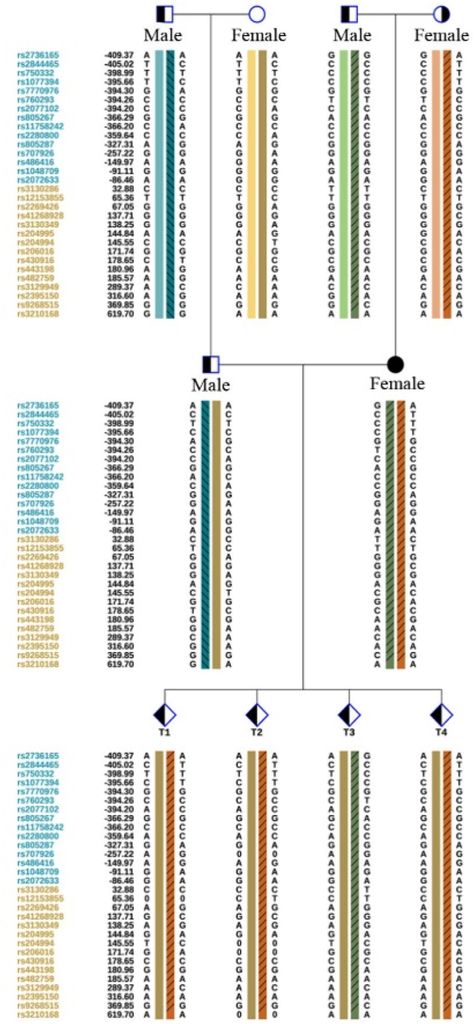
Figure 4: Detection of CYP21A2 gene mutation site and CYP21A1P/A2 fusion gene source in each embryo by WGS + SNP. The embryos T1, T2 and T4 all carried CYP21A1P/A2 fusion gene (CH-8).
The Sanger sequencing revealed a CYP21A2 mutation gene in the female proband and her father, specifically c.1034T > C (p. L354S). Furthermore, the husband of the proband and her male father carried the CYP21A2 gene c.844G>T (Figure 1). MLPA was employed for the detection of the CYP21A2 fusion gene in the family, indicating that the female proband and her mother carried the CYP21A2, CYP21A1P/A2 fusion gene (CH-8), with MPLA indicating an exon1-7 deletion (Figure 2).
Additionally, trophoblast cells of the female proband’s embryos underwent detection through PGT-M. This involved assessing trophoblast cells in each embryo using Copy-Number Variations (CNVs) and high-throughput sequencing. The CYP21A2 gene mutation sites and the origin of the CYP21A1P/A2 fusion gene in each embryo were identified through Sanger sequencing, whole-genome sequencing (WGS), and single nucleotide polymorphism (SNP) analysis. The results indicated that embryos T1, T2, and T4 all carried the CYP21A1P/A2 fusion gene (CH-8), while embryo T3 carried the c.1034T>C heterozygous mutation of maternal origin (Figure 3, 4).
The CYP21A2 genetic analysis demonstrated that the proband was a heterozygote for CYP21A2 and CYP21A1P/A2 fusion genes (CH-8). Another heterozygote for the c.1034T>C (p. L354S) mutation at different sites of the same gene was also identified. Further examination of the proband’s family revealed that the pathogenic gene mutation carried by the proband’s father was CYP21A2, c.1034T>C and the mutation carried by her mother was CYP21A2 and CYP21A1P/A2 fusion gene (CH-8).
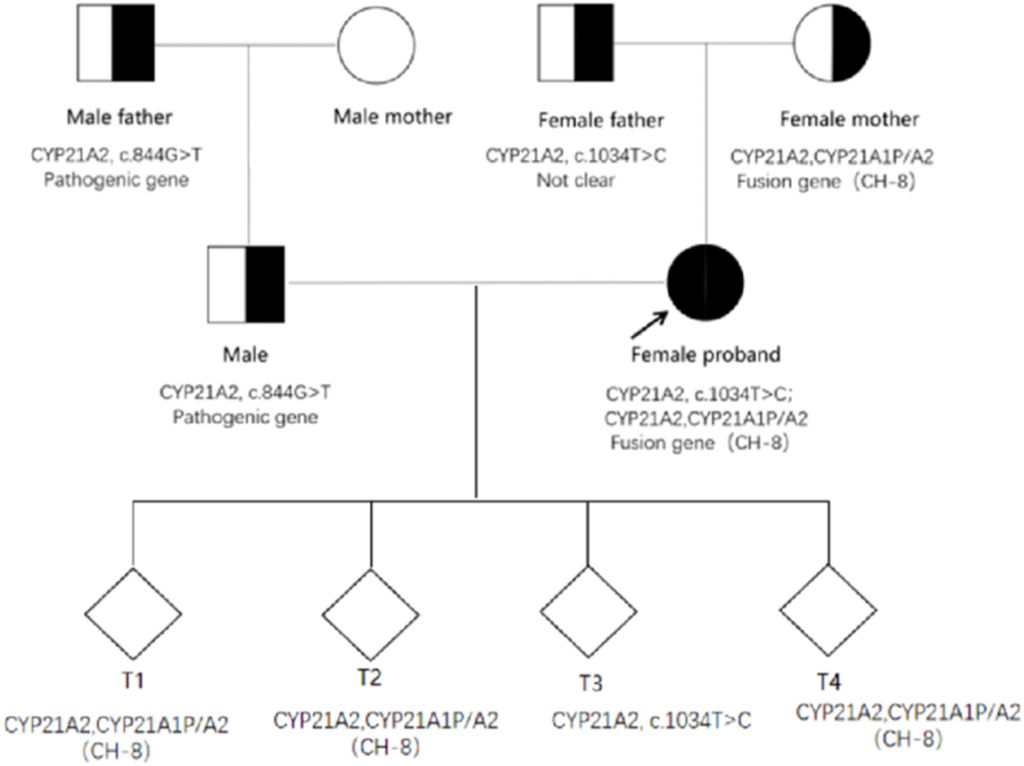
Figure 5: Family map of CYP21A2 pathogenic gene mutations.
The proband’s husband was found to have a genetic family history of related diseases. The pathogenic gene carried by this male patient and his father was CYP21A2, c.844G>T, with no CYP21A2-related gene mutation detected in his mother (Figure 5).
Due to the rarity of the congenital adrenal hyperplasia (CAH) pathogenic gene family aggregation, Preimplantation Genetic Testing for Monogenetic Disease (PGTM) was performed on the four blastocysts of the proband and her spouse through In Vitro Fertilization (IVF). The results revealed that embryos T1, T2, and T4 all carried both CYP21A2 and CYP21A1P/A2 fusion genes (CH-8), while the T3 embryo carried CYP21A2 and c.1034T>C from the proband, respectively (Figure 5).
MLPA analysis revealed the deletion of exons 1-7 in CYP21A1P/A2 fusion genes, resulting in the classic type of CAH with complete loss of enzyme activity (0). A novel point mutation, c.1034T>C (p. L354S), not previously documented, was identified, and its clinical significance remains unknown. The male’s detection of the pathogenic gene mutation c.844G>T (V282 L) in CYP21A2 indicated a nonclassical type of CAH with a 20-60% reduction in enzyme activity. Despite the milder clinical symptoms in the proband and incomplete enzyme activity loss, it is speculated that the c.1034T>C mutation leads to a lesser reduction in enzyme activity.
Four thawed blastocysts survived, and PGT was conducted, revealing T3 > T4 > T1 > T2 based on sex. The transfer sequence was evaluated comprehensively, considering other clinical indicators such as embryo rating. The patient awaited PGT results and received glucocorticoid treatment but surprisingly achieved a natural pregnancy. Currently, she is orally treated with prednisone acetate at 32 weeks of intrauterine pregnancy, showing continued progress. Noninvasive DNA testing confirmed a normal fetus. The patient declined further amniocentesis or umbilical cord blood puncture for fetal genetic testing and is currently under close follow-up.
Discussion
21OHD CAH, an autosomal recessive disorder, primarily stems from 21‑hydroxylase deficiency caused by mutations in the CYP21A2 gene. A 2022 study reveals a substantial prevalence of CYP21A2 gene mutations in 16 families with CAH. The six most frequently occurring gene mutations include Intron 2 (c.293‑13A/C>G), c.844G>T, c.1019G>A, c.92C>T, c.955C>T, and c.518T>A within the CYP21A2 gene [14]. Previous research has documented over 200 mutations of the CYP21A2 gene in various studies [11].
This study extensively presents the genetic status of a three-generation Chinese family affected by 21-hydroxylase deficiency. The pathogenic genes in the family were systematically identified and traced through detailed examination. Multiple genetic tests revealed the presence of a new mutation in the CYP21A2 gene. Analyzing the correlation between the clinical manifestation and gene phenotype of this family provided insights into the characteristics of the newly identified gene mutation site.
1. Diagnosis and Differential Diagnosis of NCCAH
1.1. Diagnosing NCCAH
- Patients often exhibit masculinization and salt wasting due to excessive androgen synthesis, leading to manifestations of female masculinity and male precocious puberty.
- In clinical practice, NCCAH falls within the purview of endocrinology specialists.
- Endocrinology-related studies indicate that diagnosing 21-hydroxylase deficiency (21OHD) relies on assessing 17-hydroxyprogesterone (17OHP) elevations, indicative of a spectrum of enzymatic defects.
- Neonatal and clinically prompted screening involves 17OHP measurement, with values >200 ng/dL (6 nmol/L) suggestive of 21OHD diagnosis.
- Classic 21OHD patients typically present 17OHP concentrations exceeding 10,000 ng/dL.
- A cosyntropin stimulation test is the standard examination method for patients with baseline 17OHP elevations of 200-1000 ng/dL (6–30 nmol/L), and values >1000 ng/dL confirm the diagnosis.
- In practical scenarios, a significant proportion of cases present atypical clinical symptoms, leading female patients to seek infertility diagnosis in reproductive clinics, resulting in frequent misdiagnosis or missed diagnosis in the clinical process.
1.2. The problem in this case
- Initially diagnosed with Polycystic Ovary Syndrome (PCOS) and primary infertility.
- Experienced repeated failures in ovulation induction treatment at other hospitals, necessitating in vitro fertilization (IVF).
- Persistent abnormal high progesterone status during Controlled Ovarian Stimulation (COS) raised concerns, eventually diagnosed through genetic testing, highlighting the importance of metabolic considerations.
1.3. In clinical practice
- NCCAH and PCOS are often confused due to their shared phenotypic characteristics, including hirsutism, ovulation disorders, and menstrual irregularities.
- Distinguishing between these syndromes based solely on clinical observations is challenging.
- Both NCCAH and PCOS are linked to obesity, insulin resistance, and dyslipidemia.
- After excluding diseases similar to Congenital Adrenal Hyperplasia (CAH), NCCAH screening is recommended, considering conditions related to ovulation reduction, anovulation, and androgen excess.
2. Correlation between Genotypes and Phenotypes
2.1. Clinical Significance of Genetic Mutation Types and Phenotypes in the Female Proband
- The pathogenic gene CYP21A2 and its closely related pseudogene CYP21A1P, located on chromosome 6 (6p21.3), are tandemly arranged and play a key role in 21-OHD [16].
- The formation of the CYP21A1P/A2 fusion gene, involving the non-encoding pseudogene CYP21A1P, may result in the deletion of varying lengths of the CYP21A2 gene, causing further impairment of 21-hydroxylase activity [17].
- Internationally, nine reported types of fusion genes are associated with classic CAH mutations [18]. Within the classical chimera group, six distinct linkage sites are identified as CH-1, CH-2, CH-3, CH-5, CH-6, and CH-7[19,20].
- A relatively recent mutation type, CH-8, characterized by the In2G mutation, is recognized. If the pseudogene’s connecting site occurs upstream of In2G, it is termed an “uncommon” chimera, resulting in slightly impaired 21-hydroxylase activity [21,22].
- The female proband, in this case, carried the CYP21A2, CYP21A1P/A2 fusion gene (CH-8), and CYP21A2, c.1034T>C (p. L354S). This suggests that diverse phenotypes in CAH patients may be influenced by different CYP21A1P/A2 fusion gene chimeras, maintaining partial 21-hydroxylase activity.
- Alternatively, variations in disease severity may be related to different types of gene mutations and their interactions. The severity of the disease could be influenced by the pathogenic mutation carried by another allele of the nonfusion gene, resulting in the phenotype of nonclassical Congenital Adrenal Hyperplasia (NCCAH). This NCCAH phenotype, as observed in the proband with menstrual disorders, infertility, and a continuous increase in progesterone, may be more favorable for overall body growth and development than a single and absolute clinical phenotype.
2.2. Clinical Implications of Genetic Mutation Types and Phenotypes in Other Family Members (Including Offspring)
- Due to the atypical symptoms associated with nonclassical Congenital Adrenal Hyperplasia (NCCAH), its detection rate in clinical practice may be underestimated, particularly in male patients.
- Family-based reporting is infrequent, as highlighted by Liu Lijun et al. [23], who reported a family in Henan, China, where all siblings suffered from CAH. Despite non-consanguineous marriage, both parents were heterozygous carriers of the CYP21A2 gene, leading to early-onset CAH in both sisters.
- The proband’s father carried CYP21A2, c.1034T>C, and the mother had the CYP21A2, CYP21A1P/A2 fusion gene (CH-8). The proband’s husband and father were carriers of the CAH pathogenic gene.
- As CAH is an autosomal recessive disorder, genetic testing and counseling were conducted to assess the risk of disease in Offspring. With a carrier spouse, there is a 1/2 probability of CAH in children per birth. The family was encouraged to consider in vitro fertilization (IVF) and preimplantation genetic testing (PGT) for eugenic purposes when necessary.
- The genetic testing report for this case revealed that all mutation sites in the CYP21A2 gene of the four embryos were inherited from the female proband. The male’s pathogenic gene was not transmitted to the Offspring.
- Three of the four embryos carried the same fusion gene (CH-8) mutation as the proband, and one (T3) had the c.1034-point mutation. Considering current research on CAH-related pathogenesis genes, using the T3 embryo for transplantation is recommended.
- The patient, who became pregnant naturally and declined fetal genetic testing due to personal preferences, necessitates long-term follow-up of the newborn. Routine examination of the newborn’s external genitalia is crucial. Deformities or abnormalities such as clitoris or penis hypertrophy should raise suspicion. If the sex is challenging to distinguish, or if abnormalities in areola and pigmentation are present, the disease should be highly suspected.
- If the newborn is female, genetic testing is recommended for both the newborn and her spouse. Relevant guidance for subsequent childbirth should be provided. The clinical significance of CYP21A2 c.1034T>C is further supported by ongoing incidence monitoring and genetic testing of the Offspring.
Conclusion
In conclusion, this family report provides a comprehensive genetic mapping of the proband and her family, elucidating the intricate genetic processes associated with various pathogenic genes and their correlation with distinct phenotypes in the patient. The study suggests that Congenital Adrenal Hyperplasia (CAH) development may be attributed to diverse mutations in the CYP21A2 gene and their interactions, influencing the varied phenotypes observed in CAH patients. However, the direct correlation between a specific individual’s phenotype and the CYP21A2 mutation remains uncertain. Future research should focus on reporting additional disease-related findings. In clinical practice, it is crucial to stay vigilant in diagnosing and treating CAH patients with multiple gene mutations, aiming for early identification to prevent misdiagnosis and overlooked cases, particularly in instances of nonclassical Congenital Adrenal Hyperplasia (NCCAH) phenotypes.
References
- Hou, Y., Li, Y., Ai, J., & Tian, L. (2024). Rare nonclassic type of Congenital adrenal hyperplasia due to 21-hydroxylase deficiency and genotype-phenotypic correlation. Heliyon, e27042. https://doi.org/10.1016/j.heliyon.2024.e27042
- Chatziaggelou A, Sakkas EG, Votino R, Papagianni M, Mastorakos G. Assisted Reproduction in Congenital Adrenal Hyperplasia. Front Endocrinol (Lausanne). 10(723) (2019) 1-6. Doi:10.3389/fendo.2019.00723
- Turcu AF, Auchus RJ. Adrenal steroidogenesis and ongenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 44 (2015) 275–96. doi: 10.1016/j.ecl.2015.02.002.
- El-maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. 390 (2017) 2194–210. doi: 10.1016/S0140-6736(17)31431-9.
- Sharma R, Seth A. Congenital adrenal hyperplasia: issues in diagnosis and treatment in children. Indian J Pediatr. 81 (2014) 178–85. doi: 10.1007/s12098-013-1280-8.
- Georgios Papadakis, Eleni A. Kandaraki, Ermioni Tseniklidi, Olga Papalou and Evanthia DiamantiKandarakis. Polycystic Ovary Syndrome and NC-CAH: Distinct Characteristics and Common Findings. A Systematic Review. (10) (2019) 388. Doi:10.3389/fendo.2019.00388.
- Parsa AA, New MI. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. 165 (2017) 2–11. doi: 10.1016/j.jsbmb.2016.06.015.
- Sharma R, Seth A. Congenital adrenal hyperplasia: issues in diagnosis and treatment in children. Indian J Pediatr. 81 (2014) 178–85. doi: 10.1007/s12098-013-1280-8.
- Witchel SF. Congenital adrenal hyperplasia. J Pediatr Adolesc Gynecol. 30 (2017) 520–34. doi:10.1016/j.jpag.2017.04.001.
- Enrico Carmina, Didier Dewailly, Héctor F Escobar-Morreale, Fahrettin Kelestimur, Carlos Moran, Sharon Oberfield, et al. nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update focusing on adolescent and adult women. Hum Reprod Update. 23(5) (2017) 580–599. DOI:10.1093/humupd/dmx014.
- Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 37 (1985) 650–67.
- Paola Concolino, Alessandra Costella. Congenital Adrenal Hyperplasia (CAH) due to 21-Hydroxylase Deficiency: A Comprehensive Focus on 233 Pathogenic Variants of CYP21A2 Gene.Mol Diagn Ther. 22(3) (2018)261-280. doi: 10.1007/s40291-018-0319-y.
- Alan A Parsa, Maria I New. Steroid 21-hydroxylase deficiency in congenital adrenal hyperplasia.J teroid Biochem Mol Biol.165(2017)2–11. doi: 10.1016/j.jsbmb.2016.06.015.
- Nils Krone, Wiebke Arlt. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 23(2) (2009)181–92. doi: 10.1016/j.beem.2008.10.014.
- Subbiah Sridhar, Ramajayam Govindhan, Balasankar Soundian, Maheshkumar Poomarimuthu, Karuppasamy Nallan, Santhanakrishnan Ramesh Kumar, et al. The Spectrum of CYP21A2 Gene Mutations from 16 Families of Congenital Adrenal Hyperplasia: Genotype-Phenotype Correlation. Indian J Endocrinol Metab.25(6) (2021)532-537. doi: 10.4103/ijem.ijem_442_21.
- Smita Jha, Adina F. Turcu. nonclassic Congenital Adrenal Hyperplasia: What Do Endocrinologists Need to Know? Endocrinol Metab Clin North Am. 50(1) (2021) 151–165. doi: 10.1016/j.ecl.2020.10.008.
- Wedell A. Congenital adrenal hyperplasia. Clin Biochem. 44(7) (2011) 505-506. DOI: 10.1016/j.clinbiochem.2011.02.026.
- Gao Yinjie, Yu Bingqing, Lu Lin, Wu Xueyan, Mao Jiangfeng, Wang Xi, et al. Analysis of copy number variation of CYP21A2 gene and the type of CYP21A1P /CYP21A2 fused gene in patients with 21⁃hydroxylase deficiency. Natl Med J China. 99(48) (2019) 3765-3769. DOI: 10.3760/cma.j.issn.0376‐2491.2019.48.002
- Chen W, Xu Z, Sullivan A, et al. Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency. Clin Chem. 58(2) (2012) 421-430. DOI:10.1373/ Clinchem.2011.174037.
- Vrzalová Z, Hrubá Z, Hrabincová ES, Vrábelová S, Votava F, Koloušková S, Fajkusová L. Chimeric CYP21A1P/CYP21A2 genes identified in Czech patients with congenital adrenal hyperplasia. Eur J Med Genet. 54 (2011) 112–7. DOI:10.1016/j.ejmg.2010.10.005.
- Lee HH. The chimeric CYP21P/CYP21 gene and 21-hydroxylase deficiency. J Hum Genet. 49 (2004) 65–72. DOI:10.1007/s10038-003-0115-2.
- White PC, Vitek A, Dupont B, New MI. Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. Proc.Natl Acad.Sci.USA. 85 (1988) 4436–40. DOI :10.1073/pnas.85.12.4436
- Wuyan Chen, Zhi Xu, Annie Sullivan, Gabriela P. Finkielstain, Carol Van Ryzin, Deborah P.Merke, and Nazli B. McDonnell. Junction Site Analysis of Chimeric CYP21A1P/CYP21A2 Genes in 21-Hydroxylase Deficiency. Clin Chem. 58(2) (2012) 421–430. doi:10.1373/clinchem.2011.174037.
- Lijun Liu, Qiang Zhang, Qi Qiao, Wenzi Li, Haijing Cui, Yanfen Ji, et al. Gene analysis of 21 hydroxylase deficiency in a sibling with congenital adrenal hyperplasia. Journal of Chinese Physician.August 20(8) (2018) 1257-1259. DOI: 10.3760/cma.j.issn.1008-1372.2018.08.045.
About Docquity
If you need more confidence and insights to boost careers in healthcare, expanding the network to other healthcare professionals to practice peer-to-peer learning might be the answer. One way to do it is by joining a social platform for healthcare professionals, such as Docquity.
Docquity is an AI-based state-of-the-art private & secure continual learning network of verified doctors, bringing you real-time knowledge from thousands of doctors worldwide. Today, Docquity has over 400,000 doctors spread across six countries in Asia. Meet experts and trusted peers across Asia where you can safely discuss clinical cases, get up-to-date insights from webinars and research journals, and earn CME/CPD credits through certified courses from Docquity Academy. All with the ease of a mobile app available on Android & iOS platforms!