Alkaptonuria, commonly called “black urine disease,” is a rare genetic condition characterized by an overproduction of homogentisic acid, which accumulate pigment polymers throughout the body. Although it is extremely rare, approximately one in every 100,000 people suffer from it. Despite the normal life expectancy, it can cause severe morbidities.
Alkaptonuria is typically managed through supportive measures such as pain relief medication, dietary adjustments, and surgical interventions, which are regarded as the gold standard of treatment. Here, we present the case of a 33-year-old man with no prior medical or surgical history who presented with severe acute back pain extending to his left leg. Genetic testing confirmed the presence of a homozygous pathogenic variant associated with Alkaptonuria.
This case underscores the difficulties in diagnosing Alkaptonuria and stresses the importance of early detection and thorough clinical assessment for better outcomes. Additionally, it highlights the need to view Alkaptonuria as a complex condition requiring a multidimensional approach, prompting further research to deepen our understanding and devise effective management strategies. Thus, this study provides a platform for future trials and investigations to delve into the complexities of Alkaptonuria, advance our knowledge, and establish comprehensive care plans for affected individuals.
Introduction on Black Urine Disease
Alkaptonuria or black urine disease is a rare autosomal recessive metabolic disorder first described in 1902 [1]. This condition arises from a deficiency in the enzyme homogentisate 1,2-dioxygenase (HGO), crucial for the breakdown of tyrosine. Consequently, homogentisic acid accumulates, forming a pigment polymer that darkens urine upon exposure to air [2]. This pigment can gradually deposit throughout the body, particularly in cartilaginous tissues, a process that unfolds over years and typically manifests in adulthood, often starting in the third decade. While some early signs may appear in childhood, such as blackish diaper staining and darkened urine, the condition is typically asymptomatic [3].
Alkaptonuria is inherited in an autosomal recessive manner, with equal chances of occurrence across genders. However, males often experience earlier and more severe clinical manifestations [4]. Patients with Alkaptonuria commonly suffer from debilitating arthritic symptoms and back pain, necessitating medical intervention [3]. Pigment deposition in heart valves can also lead to valvular calcification and cardiac complications [5].
Diagnosis of Alkaptonuria involves identifying homogentisic acid in urine through gas chromatography, while imaging studies showing disc degeneration and calcification can aid in diagnosis. Valvular involvement can be assessed using chest X-ray or CT scans, and histological evaluation of cartilage samples reveals pigment deposition outside the cells.
Management of Alkaptonuria typically involves dietary modifications and surgical interventions. Surgical options may include removing and fusing affected discs or joint replacement as necessary [6].
Case Presentation
- Patient Background: A 33-year-old male without prior medical or surgical history presented with severe acute back pain extending to his left leg. The onset of symptoms occurred two days post-heavy lifting, progressively worsening.
- Initial Assessment: Apart from weakness and numbness in the left leg, the initial evaluation highlighted signs indicative of cauda equina syndrome, prompting an urgent lumbar MRI.
- MRI Findings: The MRI unveiled a paracentral left disc herniation at the L5-S1 level, coupled with Modic type 1 changes on the inferior endplate of the L5 body (Figure 1).
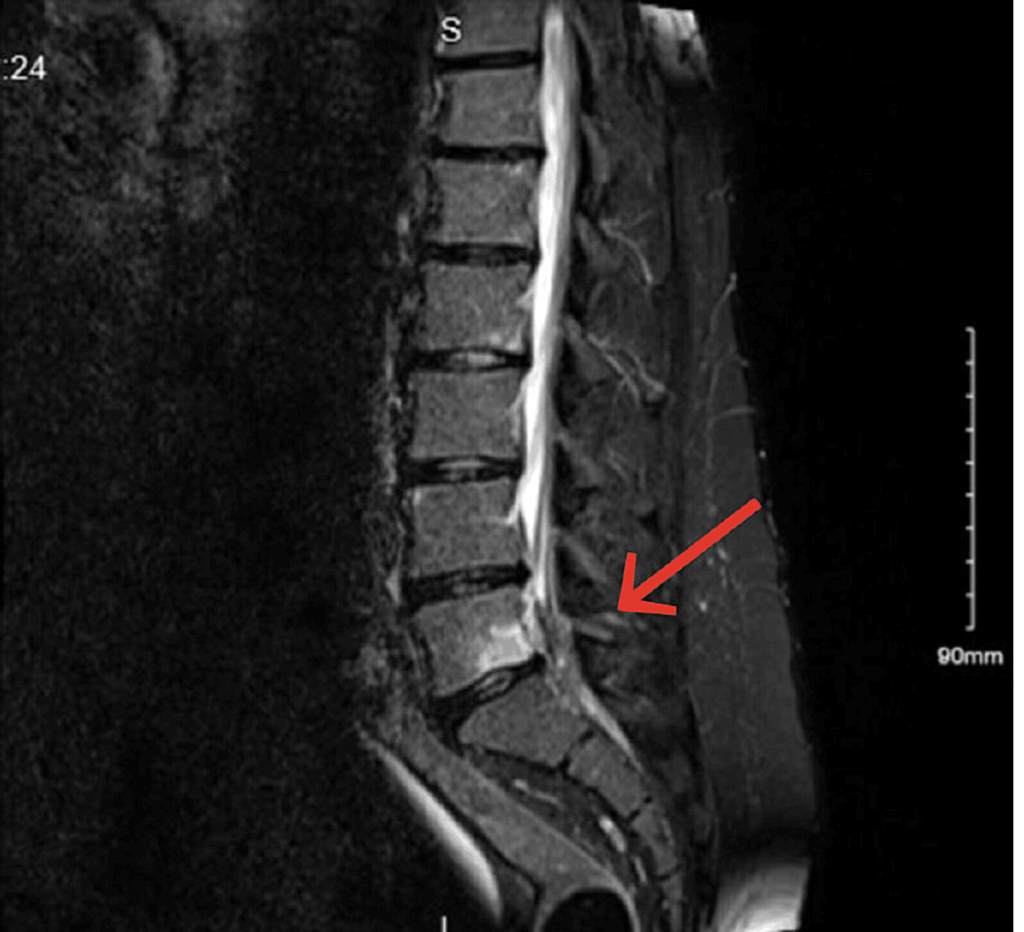
Figure 1: MRI revealed a paracentral left disc herniation at the L5-S1 level, along with Modic type 1 changes on the inferior endplate of the L5 body.
- CT Scan Evaluation: Further assessment through a CT scan aimed to identify any bone-related abnormalities contributing to compression, confirming the absence of bony osteophytes (Figure 2).
- Treatment Initiation: The patient commenced intravenous analgesia and oral pain medications.
- Reassessment: Subsequent reassessment noted persistent weakness at the L5-S1 level, graded at 3/5, alongside reduced sensation and radiating pain throughout the entire leg.
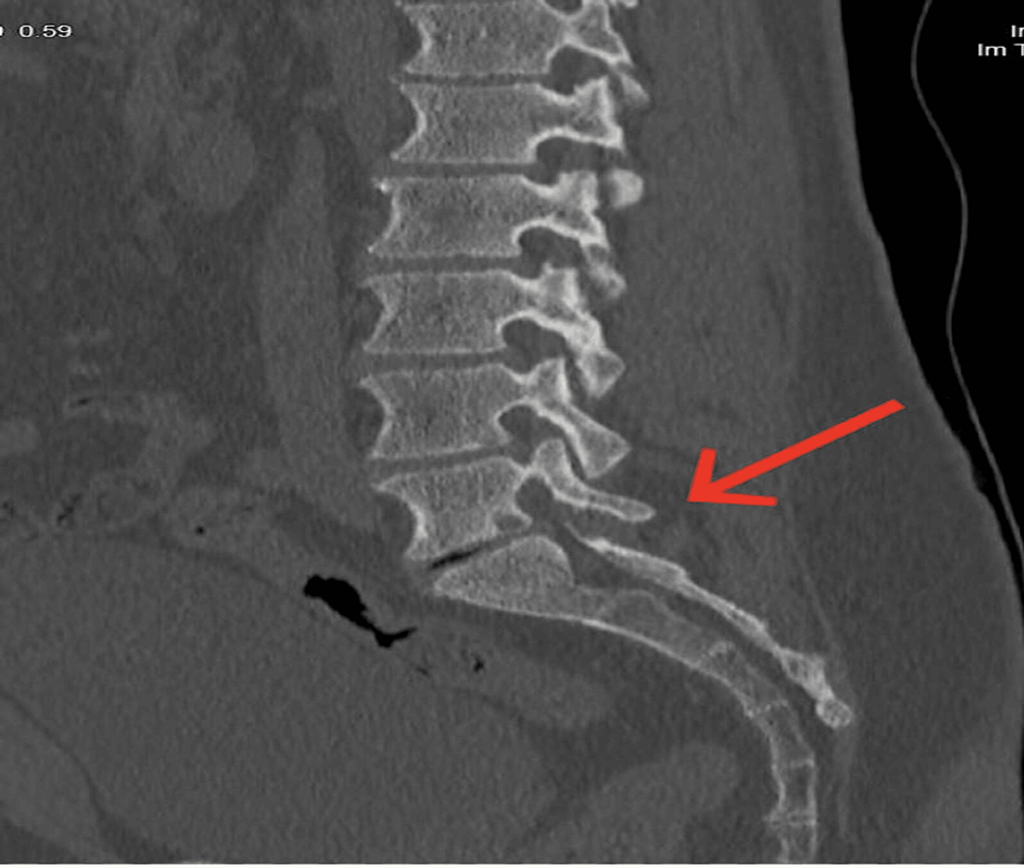
Figure 2: CT scan confirmed the absence of bony osteophytes.
- Treatment Decision: Consequently, a decision was made to proceed with a microscopic discectomy of the left L5-S1 disc the following day.
- Surgical Approach: The surgical technique adopted an open approach utilizing a minimally invasive posterior technique, with no anomalies observed in the skin, connective tissues, or bone.
- Intraoperative Observations: Upon incising the annulus fibrosus, the disc material displayed a black and dark appearance during surgery.
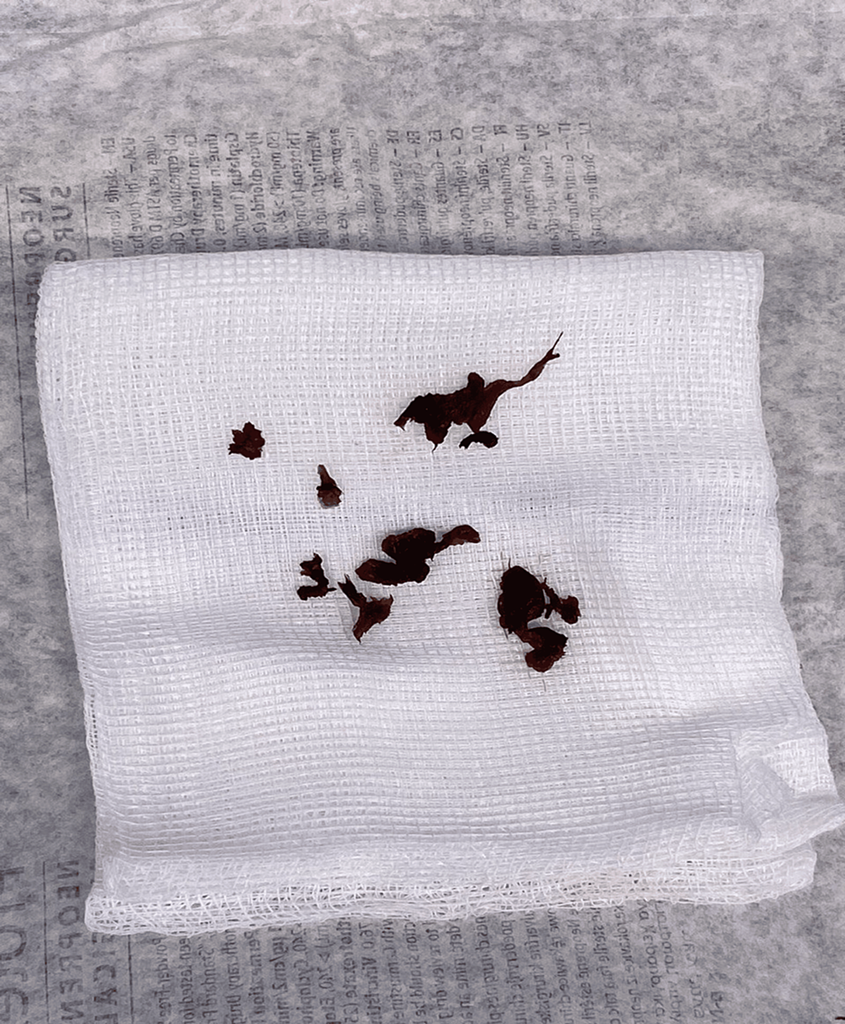
Figure 3: Intraoperative picture of the extracted disc revealed fragile and black tissues.
- Post-Surgical Examination: After removing all loose disc material, the extracted disc was inspected, revealing fragile and black tissues (Figure 3).
- Post-Surgery Outcome: Following the surgical intervention, the patient demonstrated a favorable outcome characterized by regained strength and notable alleviation of pain and numbness.
- Retrospective Evaluation: A retrospective review of the patient’s medical history and physical examination unveiled reports of varying degrees of dark urine. Notably, no pigmentation was evident in the cartilage or sclera.
- Family History: The patient disclosed having a sibling in their 30s exhibiting similar complaints of back pain and disc disease, prompting genetic testing on the patient’s siblings.
- Genetic Testing Results: The genetic analysis revealed a positive finding of a homozygous pathogenic variant in the HGD gene, confirming the diagnosis of autosomal recessive Alkaptonuria in the patient and their siblings.
- Histopathological Examination: Histopathological testing was conducted to confirm disease-related cellular or tissue changes, revealing hyperkeratosis, hypergranulosis, and evidence of fibroelastic degeneration of collagen in the affected tissues.
- Further Investigations: Urine homogentisic acid analysis via gas chromatography showed normal urine color. Additionally, an echocardiogram was conducted to rule out valve abnormalities associated with Alkaptonuria, yielding negative results.
- Recovery Progression: The patient’s recovery progressed smoothly without any complications.
- One-Year Follow-Up: After a year of follow-up, the patient displayed no symptoms or signs of the previously diagnosed disease, indicating complete resolution or remission.
- Significance of Follow-Up: The follow-up period provided reassurance regarding the patient’s well-being and facilitated a comprehensive assessment of their long-term health status.
- Clinical Implications: The positive clinical outcome, coupled with genetic and histopathological findings, offers valuable insights into the nature and potential hereditary aspects of the disease, contributing to enhanced diagnosis, treatment, and genetic counseling for the patient and their family members.
Discussion
Initially documented by Garrod in 1902 as an autosomal recessive genetic disorder, Alkaptonuria is characterized by a deficiency in the HGD enzyme, leading to the accumulation of homogentisic acid in connective tissues [1]. Chromosome 3 has been identified as the location of the defective gene [1]. This buildup of homogentisic acid oxidizes into a melanin-like polymer, resulting in the distinctive dark yellow pigmentation observed in cartilage and other tissues [1,7]. Typically, patients remain asymptomatic until their third or fourth decades, with early signs manifesting as darkened urine upon standing [8]. By age 40, individuals may exhibit external signs of ochronosis, characterized by bluish-black discoloration, with pigmentation often more prominent in sun-exposed areas, cartilage, and regions with high sweat gland density [9]. Approximately 50% of patients develop ochronotic arthropathy, a musculoskeletal manifestation of Alkaptonuria, which affects weight-bearing joints and can lead to arthritis and significant back pain [10,11]. Common symptoms include back stiffness, lordosis loss, ly thoracic kyphosis, intervertebral calcification, and vacuum phenomena with radiolucent gas collections frequently observed on radiological imaging [12]. While Alkaptonuria affects both genders equally, males typically experience more severe and earlier-onset arthritic symptoms [13]. Diagnosis is often delayed until intraoperative identification during orthopedic surgery, owing to the condition’s rarity and its symptomatic similarity to other forms of arthritis. There is no definitive treatment for Alkaptonuria; however, management strategies such as a low tyrosine and phenylalanine diet, physical therapy, NSAIDs, and antioxidants are commonly recommended [1,10].
Conclusion
Alkaptonuria is often overlooked in clinical practice, highlighting the need for enhanced understanding and early detection. Identifying this condition promptly is vital for preventing and managing its impact on various bodily systems, encompassing skin changes and renal, cardiovascular, and hepatic involvement. Hence, it is imperative to heighten awareness among healthcare professionals regarding the significance of evaluating Alkaptonuria in unexplained musculoskeletal symptoms, given its frequent association with such presentations. With no effective treatment currently available, a comprehensive approach to addressing Alkaptonuria as a multifaceted disorder is warranted, underscoring the importance of further research. This study offers a platform for conducting trials and series to deepen our understanding of Alkaptonuria and its complexities.
References
- Alhelal F, Alissa S, Abaalkhail M, et al. (October 07, 2023) Alkaptonuria Diagnosis Following a Discectomy: A Case Report. Cureus 15(10): e46644. doi:10.7759/cureus.46644.
- Garrod AE: The incidence of Alkaptonuria: a study in chemical individuality. 1902. Mol Med. 1996, 2:274-82.
- Lodh M, Kerketta JA: Early diagnosis of co-existent ß-thalassemia and Alkaptonuria. Indian J Hum Genet. 2013, 19:259-61. 10.4103/0971-6866.116104
- Ranganath LR, Norman BP, Gallagher JA: Ochronotic pigmentation is caused by homogentisic acid and is the key event in Alkaptonuria leading to the destructive consequences of the disease – a review. J Inherit Metab Dis. 2019, 42:776-92. 10.1002/jimd.12152
- Craide FH, Fonseca JS, Mariano PC, Fernandez NM, Castro CG, Mene Y: Alkaptonuria – case report. An Bras Dermatol. 2014, 89:799-801. 10.1590/abd1806-4841.20143052
- Lok ZS, Goldstein J, Smith JA: Alkaptonuria-associated aortic stenosis. J Card Surg. 2013, 28:417-20. 10.1111/jocs.12129
- Mistry JB, Bukhari M, Taylor AM: Alkaptonuria. Rare Dis. 2013, 1:10.4161/rdis.27475
- Dogra A, Bajwa GS, Bajwa N, Khurana S: Alkaptonuria. Indian J Dermatol Venereol Leprol. 2001, 67:271-2.
- Verma SB: Early detection of Alkaptonuria. Indian J Dermatol Venereol Leprol. 2005, 71:189-91. 10.4103/0378-6323.16236
- Mannoni A, Selvi E, Lorenzini S, et al.: Alkaptonuria, ochronosis, and ochronotic arthropathy. Semin Arthritis Rheum. 2004, 33:239-48. 10.1053/s0049-0172(03)00080-5
- Du Jr BN: Alcaptonuria and ochronotic arthritis. Mol Biol Med. 1991, 8:31-8.
- Damarla N, Linga P, Goyal M, Tadisina SR, Reddy GS, Bommisetti H: Alkaptonuria: a case report. Indian J Ophthalmol. 2017, 65:518-21. 10.4103/ijo.IJO_337_16
- Khan AH, Afroze B, Majid H, Zaidi Y, Jamil A, Jafri L: Musculoskeletal manifestations in Alkaptonuria: a cross-sectional study. Medicine (Baltimore). 2021, 100:10.1097/MD.0000000000028241
- Wu K, Bauer E, Myung G, Fang MA: Musculoskeletal manifestations of Alkaptonuria: a case report and literature review. Eur J Rheumatol. 2019, 6:98-101. 10.5152/eurjrheum.2018.18116
About Docquity
If you need more confidence and insights to boost careers in healthcare, expanding the network to other healthcare professionals to practice peer-to-peer learning might be the answer. One way to do it is by joining a social platform for healthcare professionals, such as Docquity.
Docquity is an AI-based state-of-the-art private & secure continual learning network of verified doctors, bringing you real-time knowledge from thousands of doctors worldwide. Today, Docquity has over 400,000 doctors spread across six countries in Asia. Meet experts and trusted peers across Asia where you can safely discuss clinical cases, get up-to-date insights from webinars and research journals, and earn CME/CPD credits through certified courses from Docquity Academy. All with the ease of a mobile app available on Android & iOS platforms!